
As filed with the U.S. Securities and Exchange Commission on November 19, 2018
Registration No. 333-228313
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
_______________________
Amendment No. 1 to
FORM S-1
REGISTRATION STATEMENT UNDER THE SECURITIES ACT OF 1933
_______________________
DIAMEDICA THERAPEUTICS INC.
(Exact name of registrant as specified in its charter)
|
Canada (State or other jurisdiction of incorporation or organization) |
2834 (Primary Standard Industrial Classification Code Number) |
Not Applicable (I.R.S. Employer Identification Number) |
2 Carlson Parkway, Suite 260
Minneapolis, Minnesota 55447
(763) 496-5454
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
_______________________
Rick Pauls
President and Chief Executive Officer
DiaMedica Therapeutics Inc.
2 Carlson Parkway, Suite 260
Minneapolis, Minnesota 55447
(763) 496-5454
(Name, address, including zip code, and telephone number, including area code, of agent for service)
_______________________
Copies to:
|
Amy E. Culbert Brett R. Hanson Fox Rothschild LLP Campbell Mithun Tower, Suite 2000 222 South Ninth Street Minneapolis, Minnesota 55402 (612) 607-7000 |
Keith Inman Pushor Mitchell LLP 301 – 1665 Ellis Street Kelowna, British Columbia Canada V1Y 2B3 (250) 762-2108 |
Jonathan R. Zimmerman Ben A. Stacke Faegre Baker Daniels LLP 2200 Wells Fargo Center 90 South Seventh Street Minneapolis, Minnesota 55402 (612) 766-7000 |
Approximate date of commencement of proposed sale to the public: As soon as practicable after the effective date of this registration statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box: ☐
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration for the same offering. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer ☐ |
Accelerated filer ☐ |
|
Non-accelerated filer ☐ |
Smaller reporting company ☒ |
|
Emerging growth company ☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act. ☒
CALCULATION OF REGISTRATION FEE
|
Title of Each Class of Securities to be Registered(1) |
Proposed Maximum Aggregate Offering Price(2) |
Amount of Registration Fee(3)(4) |
|
Voting Common Shares, no par value per share |
$15,000,000 |
$1,818 |
|
(1) |
Each voting common share, no par value per share (“common shares”), includes one common share purchase right pursuant to a Shareholder Rights Plan Agreement dated December 21, 2017 by and between DiaMedica Therapeutics Inc. and Computershare Investor Services Inc. Any value attributable to such rights is reflected in the market price of the common shares. |
|
(2) |
Estimated solely for purposes of calculating the amount of the registration fee pursuant to Rule 457(o) under the Securities Act of 1933, as amended. Includes the offering price of shares that the underwriter has the option to purchase to cover over-allotments, if any. |
|
(3) |
Calculated pursuant to Rule 457(o) based on an estimate of the proposed maximum aggregate offering price. |
|
(4) |
Previously paid. |
_______________________
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the registration statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED NOVEMBER 19, 2018
PRELIMINARY PROSPECTUS
Shares
Common Shares
_______________________
This is the initial public offering of common shares of DiaMedica Therapeutics Inc. in the United States. All common shares being offered are being sold by the Company. We expect the initial public offering price will be between $ and $ per share. We have applied to list our common shares in the United States on The Nasdaq Capital Market under the symbol “DMAC.” Although this is our initial public offering of our common shares in the United States, our common shares trade in Canada on the TSX Venture Exchange under the symbol “DMA” and over-the-counter in the United States on the OTCQB marketplace under the symbol “DMCAF.” The last reported sale price of our common shares at the close of business on November 16, 2018 on the TSX Venture Exchange was CAD $6.50 per share (US $4.94) and over-the-counter in the United States as quoted by the OTCQB marketplace was $4.95 per share.
We are an “emerging growth company” under applicable Securities and Exchange Commission rules and will be eligible for, and have decided to comply with, reduced public company disclosure requirements in future reports after completion of this offering. See “Prospectus Summary – Implications of Being an Emerging Growth Company.”
Investing in our common shares involves a high degree of risk. See “Risk Factors” beginning on page 11 of this prospectus for a discussion of information that should be considered in connection with an investment in our common shares.
_______________________
Neither the Securities and Exchange Commission nor any state securities regulators have approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
_______________________
|
Per Share |
Total |
|||||||
|
Initial public offering price |
$ | $ | ||||||
|
Underwriting discounts and commissions(1) |
$ | $ | ||||||
|
Proceeds, before expenses, to us |
$ | $ | ||||||
__________________________
|
(1) |
In addition, we have agreed to reimburse the underwriter for certain expenses. See “Underwriting” for additional disclosure regarding underwriting discounts, commissions and estimated offering expenses. |
We have granted the underwriter an option for a period of 30 days from the date of this prospectus to purchase up to an additional common shares from us at the initial public offering price less the underwriting discounts and commissions to cover over-allotments, if any.
Delivery of the common shares is expected to be made on or about , 2018, subject to customary closing conditions.
___________________
Craig-Hallum Capital Group
Prospectus dated , 2018
TABLE OF CONTENTS
|
|
Page |
|
Prospectus Summary |
1 |
|
Summary Consolidated Financial Data |
10 |
|
Risk Factors |
11 |
|
Special Note Regarding Forward-Looking Statements |
44 |
|
Use of Proceeds |
45 |
|
Price Range of Our Common Shares |
46 |
|
Dividend Policy |
47 |
|
Capitalization |
48 |
|
Dilution |
49 |
|
Selected Financial Data |
50 |
|
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
51 |
|
Business |
59 |
|
Management |
80 |
|
Executive and Director Compensation |
88 |
|
Certain Relationships and Related Party Transactions |
97 |
|
Principal Shareholders |
99 |
|
Description of Share Capital |
101 |
|
Shares Eligible For Future Sale |
112 |
|
Certain United States Federal Income Tax Considerations |
114 |
|
Material Canadian Federal Income Tax Considerations |
120 |
|
Underwriting |
121 |
|
Legal Matters |
127 |
|
Experts |
127 |
|
Where You Can Find Additional Information |
127 |
|
Index To Consolidated Financial Statements |
F-1 |
ABOUT THIS PROSPECTUS
You should rely only on the information contained in this prospectus and any free writing prospectus prepared by or on behalf of us or to which we have referred you. Neither we nor the underwriter has authorized anyone to provide you with information that is different. We are offering to sell our common shares, and seeking offers to buy our common shares, only in jurisdictions where offers and sales are permitted. The information in this prospectus is complete and accurate only as of the date on the front cover of this prospectus, regardless of the time of delivery of this prospectus or any sale of our common shares.
For investors outside the United States: neither we nor the underwriter have done anything that would permit this offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus must inform themselves about, and observe any restrictions relating to, the offering of our common shares and the distribution of this prospectus outside the United States.
Except as otherwise indicated herein or as the context otherwise requires, references in this prospectus to “DiaMedica,” “the Company,” “we,” “us,” “our” or similar references mean DiaMedica Therapeutics Inc. and its subsidiaries. References in this prospectus to “common shares” means our voting common shares, no par value per share.
We own various unregistered trademarks and service marks, including our corporate logo. Solely for convenience, the trademarks and trade names in this prospectus are referred to without the ® and ™ symbols, but such references should not be construed as any indicator that the owner of such trademarks and trade names will not assert, to the fullest extent under applicable law, their rights thereto. We do not intend the use or display of other companies’ trademarks and trade names to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
INDUSTRY AND MARKET DATA
In addition to the industry, market and competitive position data referenced in this prospectus from our own internal estimates and research, some market data and other statistical information included in this prospectus are based in part upon information obtained from third-party industry publications, research, surveys and studies, none of which we commissioned. Third-party industry publications, research, surveys and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information.
We are responsible for all of the disclosure in this prospectus and while we believe that each of the publications, research, surveys and studies included in this prospectus are prepared by reputable sources, neither we nor the underwriter have independently verified market and industry data from third-party sources. In addition, while we believe our internal company research and estimates are reliable, such research and estimates have not been verified by independent sources. Assumptions and estimates of our and our industry’s future performance are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in “Risk Factors.” These and other factors could cause our future performance to differ materially from our assumptions and estimates. See “Special Note Regarding Forward-Looking Statements.”
PROSPECTUS SUMMARY
The following summary highlights information contained elsewhere in this prospectus and is qualified in its entirety by the more detailed information and financial statements included elsewhere in this prospectus. This summary does not contain all of the information you should consider before investing in our common shares. Before you decide to invest in our common shares, you should read and carefully consider the following summary together with the entire prospectus, including our financial statements and the related notes thereto appearing elsewhere in this prospectus and the matters discussed in the sections in this prospectus entitled “Risk Factors,” “Selected Financial Data” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” Some of the statements in this prospectus constitute forward-looking statements that involve risks and uncertainties. See “Special Note Regarding Forward-Looking Statements.” Our actual results could differ materially from those anticipated in such forward-looking statements as a result of certain factors, including those discussed in the “Risk Factors” and other sections of this prospectus.
Overview
We are a clinical stage biopharmaceutical company primarily focused on the development of novel recombinant (synthetic) proteins. Our goal is to use our patented and licensed technologies to establish our company as a leader in the development and commercialization of novel recombinant proteins to treat neurological and kidney diseases. Our primary focus is on acute ischemic stroke (“AIS”) and chronic kidney disease (“CKD”). We plan to advance our lead drug candidate, DM199, through clinical trials, as appropriate, to create shareholder value by establishing its clinical and commercial potential as a therapy for AIS and CKD.
DM199 is a recombinant form of human tissue kallikrein-1 (“KLK1”). KLK1 is an endogenous serine protease (protein) produced in the kidneys, pancreas and salivary glands, which plays a critical role in the regulation of local blood flow and vasodilation (the widening of blood vessels which decreases blood pressure) in the body, as well as an important role in managing inflammation and oxidative stress (an imbalance between potentially damaging reactive oxygen species, or free radicals, and antioxidants in your body). We believe DM199 has the potential to treat a variety of diseases where healthy functioning requires sufficient activity of KLK1 and its biochemical system, the kallikrein-kinin system (“KKS”).
AIS and CKD patients suffer from a lack of blood flow to the brain and kidneys, respectively. These patients also tend to exhibit lower than normal levels of endogenous KLK1. We believe treatment with DM199 could replenish low levels of endogenous KLK1, thereby releasing physiological levels of bradykinin (“BK”) when and where needed, generating beneficial nitric oxide and prostacyclin setting in motion metabolic pathways that can improve blood flow (through vasoregulation) to damaged end-organs, such as the brain and kidneys, supporting the structural integrity and normal functioning.
Today, forms of KLK1 derived from human urine and porcine pancreas are sold in Japan, China and Korea to treat AIS, CKD, retinopathy, hypertension and related vascular diseases. We believe millions of patients have been treated with these KLK1 therapies, and the data from more than 100 published papers and studies support their clinical benefit. However, there are numerous regulatory, commercial, and clinical drawbacks associated with KLK1 derived from human urine and porcine pancreas that can be overcome by developing a synthetic version of KLK1 such as DM199. We believe regulatory drawbacks are the primary reason why KLK1 derived from human urine and porcine pancreas are not currently available and used in the United State or Europe. We are not aware of any synthetic version of KLK1 with regulatory approval for human use in any country, nor are we aware of any synthetic version in development other than our drug candidate DM199. We believe at least five companies have attempted to create a synthetic version of KLK1, but have been unsuccessful.
We have conducted numerous internal and third-party analyses to evaluate the structural and functional performance of DM199 as compared to KLK1 derived from human urine. The results of these studies have demonstrated that DM199 is structurally and functionally equivalent to KLK1 derived from human urine in that (i) the amino acid structure of DM199 is identical to the human urine form, (ii) the enzymatic and pharmacokinetic profiles are substantially similar to human urinary derived KLK1 and (iii) the physiological effects of DM199 on blood pressure mirror that of human urinary derived KLK1. We believe that the results of this work suggest that the therapeutic action of DM199 will be the same or better than that of the forms of KLK1 marketed in Asia. In addition, we have completed five clinical trials with DM199 treating over 120 volunteers and the results have shown that DM199 has been well-tolerated. However, DM199 has not been, and we cannot provide any assurance that it ultimately will be, determined to be safe or effective for purposes of granting marketing approval by the FDA or any comparable agency.
1
Our recombinant form of DM199 is protected by issued composition of matter and delivery patents in the United States and Europe (2033); a pending worldwide patent (2038) that covers a range of DM199 dose levels and dosing regimens useful for treating a wide range of diseases associated with microvascular dysfunction; an exclusive license with our manufacturing partner for use of their cell line and proprietary expression system for manufacturing synthetic KLK1; and numerous trade-secrets. In addition, we believe DM199 cannot be reverse engineered to develop a copycat version of our therapy. This adds additional protection to our intellectual property, especially as we engage in DM199 licensing activities. We do not intend to share formulary secrets with licensing partners outside of the United States, but we will supply our partners with bulk active ingredient which we manufacture domestically.
Our Programs
The primary focus for our DM199 program development is on AIS and CKD; however, we also intend to pursue advancement in the vascular dementia market. The current status of our product candidates in clinical development is as follows:

|
● |
Acute Ischemic Stroke. According to the World Health Organization, each year approximately 15 million people worldwide suffer a stroke, of which 5.0 million will die and 5.0 million will be permanently disabled. The majority of stroke patients suffer an ischemic event, which according to the U.S. Center for Disease Control and Prevention is approximately 87% of all stroke patients. We believe that stroke represents an area of significant unmet medical need, and a KLK1 treatment (such as DM199) could provide a significant patient benefit with its proposed therapeutic window of up to 24 hours after the first sign of symptoms. Currently, the only FDA-approved pharmacological intervention for AIS is tissue plasminogen activator (“tPA”), which must be given within 4.5 hours of symptom onset. Treating patients with tPA during this time window can be challenging because it is difficult to determine precisely when symptoms began and a patient must undergo complex brain imaging before treatment to rule out a hemorrhagic stroke. Mechanical thrombectomy, a procedure in which the clot is removed using catheter-based tools, is also available to certain patients. Despite the availability of these treatments, we believe they are relevant to less than 10% of ischemic stroke patients due to the location of the clot, the elapsed time after the stroke occurred, or safety considerations. Thus, we believe DM199 may offer significant advantages over the current treatment options and fill an unmet need for patients who cannot receive tPA or mechanical thrombectomy (most stroke patients will be diagnosed in less than 24 hours). Additionally, DM199 may also offer a complimentary follow-on treatment for patients who initially receive tPA or mechanical thrombectomy treatments by enabling sustained blood flow improvements to the brain during the critical first few weeks after a stroke. Based on the number of strokes each year (approximately 1.7 million in the U.S., Europe and Japan and 15 million worldwide) and the $8,500 estimated cost per patient for the current standard of care, tPA, we believe the annual market opportunity for DM199 could be significant. |
2
|
● |
Chronic Kidney Disease. CKD is a widespread health problem that generates significant economic burden throughout the world. According to the National Kidney Foundation, 30 million Americans and 120 million Chinese suffer from this debilitating and potentially life-threatening condition. CKD is a progressive condition causing the kidneys to lose function over time, increasing the risk of premature death, cardiovascular events, and hospitalization. End stage renal disease (“ESRD”) is the final stage of CKD and requires ongoing dialysis or a kidney transplant to survive, but many patients suffer serious health consequences or die from CKD prior to developing ESRD. Currently, there is no cure for CKD and treatment involves management of the disease. Blood pressure medications, such as angiotensin converting enzyme inhibitors (“ACEi”) or angiotensin receptor blockers (“ARB”), are often prescribed to control hypertension, and hopefully, slow the progression of CKD. Nevertheless, according to the National Kidney Foundation, many patients continue to show declining kidney function. We believe DM199 offers a potentially novel approach for the treatment of CKD because KLK1 protein plays a vital role in normal kidney function. Since patients with moderate to severe CKD often excrete abnormally low levels of KLK1 in their urine, we believe that DM199 may prevent or reduce further kidney damage by replenishing endogenous KLK1 and restoring the protective kallikrein-kinin system to regulate the production and release of nitric oxide and prostacyclin. |
Human Urine-Extracted KLK1 Studies in AIS Patients
In China, a human urine-extracted KLKl protein (Kailikang®) is approved and marketed by Techpool Bio-Pharma Inc., a company controlled by Shanghai Pharmaceuticals Holding Co. Ltd. According to a publication in the China Journal of Neurology, in a double-blinded, placebo-controlled trial of 446 patients treated with either KLK1 or a placebo administered up to 48 hours after a stroke, such patients showed significantly better scores on the European Stroke Scale and Activities of Daily Living at three weeks post-treatment and after three months using the Barthel Index. Numerous internal and Third party analyses demonstrate DM199 bioequivalence to Kailikang®.
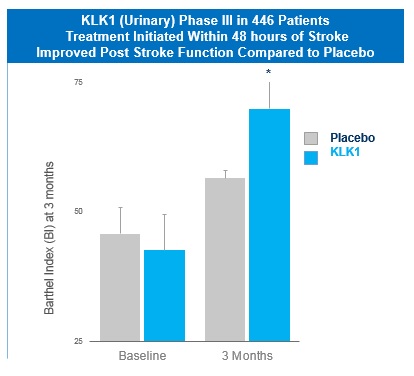
Furthermore, a comprehensive meta-analysis covering 24 clinical studies involving 2,433 patients published in the Journal of Evidenced Based Medicine concluded that human urinary KLK1 appears to ameliorate neurological deficits for patients with AIS and improves long-term outcomes, though a few treated patients suffered from transient hypotension.
3
Porcine-Derived KLK1 Studies in CKD Patients
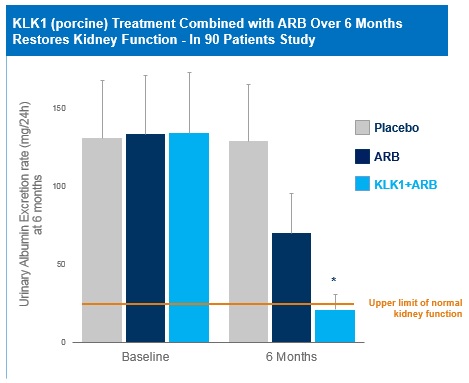
Over 20 clinical papers have been published in the Chinese literature supporting the therapeutic activity in CKD patients of porcine KLK1 given alone or in combination with an ARB or an ACEi. These unblinded studies involve treatment durations ranging from a few weeks up to six months and report improvement in kidney disease based on decreased urinary albumin excretion rates (“UAER”) and other clinical endpoints of kidney disease. In a 2011 study of 90 patients, CKD patients treated with porcine KLK1 combined with ARB restored kidney function to normalized levels based on UAER.
License to Ahon Pharma, a Subsidiary of Fosun Pharma
On September 27, 2018, we entered into a license and collaboration agreement with Ahon Pharmaceutical Co Ltd. (“Ahon Pharma”), a subsidiary of Shanghai Fosun Pharmaceutical (Group) Co. Ltd. (“Fosun Pharma”), which grants Ahon Pharma the exclusive rights to develop and commercialize DM199 for acute ischemic stroke in mainland China, Taiwan, Hong Kong S.A.R. and Macau S.A.R. Fosun Pharma is one of China’s largest pharmaceutical firms with annual sales of more than USD $2 billion and an extensive related hospital sales force. Under the terms of the license agreement, we are entitled to receive an upfront payment of $5 million, consisting of $500,000 on signing and $4.5 million upon regulatory clearance to initiate a clinical trial in China. We also have the potential to receive an additional $27.5 million in development and sales related milestones and up to approximately 10% royalties on net sales of DM199 in the licensed territories. All development, regulatory, sales, marketing, and commercial activities and associated costs in the licensed territories will be the sole responsibility of Ahon Pharma. Fosun Pharma, through its partnership with SK Group (a South Korea based Fortune Global 100 Company) is an investor in DiaMedica through an equity investment made in 2016.
Potential DM199 Commercial Advantages
The growing understanding of KLK1’s role in human health and its use in Asia as an approved therapeutic highlights two important potential commercial advantages for DM199:
|
● |
KLK1 treatments currently sold in Japan, China and Korea. Research has shown that patients with low levels of KLK1 are associated with a variety of diseases related to vascular dysfunction, such as chronic kidney disease, acute ischemic stroke, retinopathy and hypertension. Clinical trial data with human urine and porcine derived KLK1 has demonstrated statistically significant clinical benefits in treating a variety of patients with KLK1 compared to placebo. These efficacy results are further substantiated by established markets in Japan, China and Korea for pharmaceutical sales of KLK1 derived from human urine and porcine pancreas. We estimate that millions of patients have been treated with these forms of KLK1 in Asia. Altogether, we believe this supports a strong market opportunity for a synthetic version of KLK1 such as DM199. |
4
|
● |
KLK1 treatment has had limited side effects and has been well tolerated to date. KLK1 is naturally produced by the human body; and, therefore, the body’s own control mechanisms act to limit potential side effects. The only notable side effect observed in our clinical trials was orthostatic hypotension, or a sudden drop in blood pressure, which was only seen at doses ten to twenty times higher than our anticipated therapeutic dose levels. Moreover, routine clinical use of KLK1 treatment in Asia we understand has been well-tolerated by patients for several decades. In 2017, we completed a clinical trial comparing the pharmacokinetic profile of DM199 to the human urinary form of KLK1 (Kailikang®), which showed DM199, when administered in intravenous form, had a similar pharmacokinetic profile. Further, when DM199 was administered subcutaneously, DM199 demonstrated a longer acting pharmacokinetic profile, superior to the intravenously administered Kailikang® and DM199. |
In addition, we believe there are also significant formulation, manufacturing, regulatory and other advantages for our synthetic human KLK1 drug candidate DM199:
|
● |
Potency and Impurity Considerations. KLK1 derived from human urine or porcine pancreas may contain impurities, endotoxins, and chemical byproducts due to the inherent variability of the isolation and purification process. We believe that this creates the risk of inconsistencies in potency and impurities from one production run to the next. However, we expect to produce a consistent formulation of KLK1 that is free of endotoxins and other impurities. |
|
● |
Cost and Scalability. Large quantities of human urine and porcine pancreas must be obtained to derive a small amount of KLK1. This creates potential procurement, cost and logistical challenges to source the necessary raw material, particularly for human urine sourced KLK1. Once sourced, the raw material is processed using chemicals and costly capital equipment and produces a significant amount of byproduct waste. Our novel recombinant manufacturing process utilizes widely available raw materials and can be readily scaled for commercial production. Accordingly, we believe our manufacturing process will have significant cost and scalability advantages. |
|
● |
Regulatory. We are not aware of any attempts by manufacturers of the urine or porcine based KLK1 products to pursue regulatory approvals in the United States. We believe that this is related to challenges presented by using inconsistent and potentially hazardous biomaterials, such as human urine and porcine pancreas, and their resulting ability to produce a consistent drug product. Our novel recombinant manufacturing process utilizes widely available raw materials which we believe provides a significant regulatory advantage, particularly in regions such as the United States, Europe and Canada, where safety standards are high. In addition, we believe that DM199 could qualify for 12 years of data exclusivity under the Biologics Price Competition and Innovation Act of 2009, which was enacted as part of the Patient Protection and Affordable Care Act as amended by the Health Care and Education Reconciliation Act of 2010 (collectively, the “ACA”). |
Our Strategy
We aim to become a leader in the development and commercialization of novel recombinant proteins to treat neurological and kidney diseases. To achieve this goal, we are pursuing the following strategies:
|
● |
Complete our Phase II clinical development for DM199 in AIS patients; |
|
● |
Advance Phase Ib and Phase II studies for DM199 in CKD patients; |
|
● |
Initiate a Phase II study for DM199 in patients with vascular dementia; |
|
● |
Explore potential new indications for DM199; |
|
● |
Leverage our experience and technologies to develop new recombinant therapies and programs; and |
|
● |
License our lead product candidate into new territories and prepare for commercialization of DM199. |
5
Our Team
We have assembled a seasoned management team with extensive experience in drug discovery, development, manufacturing, and commercialization. Our Chief Executive Officer, Rick Pauls, MBA, is a successful venture capitalist and formerly the Co-Founder and Managing Director of CentreStone Ventures Inc., a life sciences venture capital fund which made early investments in DiaMedica. Our Chief Scientific Officer, Todd Verdoorn, PhD, has more than 26 years of experience in the pharmaceutical and biotechnology industries, including five years working with Bristol-Myers Squibb’s stroke group. Our Chief Medical Officer, Harry Alcorn Jr., PhD, has more than 30 years’ experience planning, operating, and executing clinical development programs across a range of diseases including kidney disease, diabetes, and cardiovascular disease, and most recently served as Chief Scientific Officer of DaVita Clinical Research. Our Chief Financial Officer, Scott Kellen, CPA, brings over two decades of operational and corporate finance expertise including an extensive background working with publicly-traded healthcare and biotechnology companies.
Over the last two years we have also been supported by significant investments from Hermed Capital, an investment fund affiliated with our partner Fosun Pharma’s subsidiary Ahon Pharma, and Dr. Nancy Chang, PhD, CEO and President of Apex Enterprises, and a member of our strategic advisory board.
Risks Affecting Us
Our business is subject to a number of risks of which you should be aware before making an investment decision. These risks are discussed more fully in the “Risk Factors” section of this prospectus immediately following this prospectus summary. Some of these risks include:
|
● |
we are an early stage company with no approved products and no revenue; |
|
● |
our prospects depend on the success of our DM199 product candidate; |
|
● |
we rely on third parties to plan, conduct and monitor our preclinical and clinical trials; |
|
● |
we rely on a contract manufacturer over whom we have limited control to manufacture DM199; |
|
● |
we are in litigation with a contract research organization which could harm our ability to obtain regulatory approval for DM199; |
|
● |
clinical trials are expensive and complex with uncertain outcomes, which may prevent or delay regulatory approval of or commercialization of our DM199 product candidate; |
|
● |
we may not be successful in finding collaboration partners to assist us with the development or commercialization of our DM199 product candidate; |
|
● |
regulatory approval processes are lengthy, expensive and inherently unpredictable, and even if our DM199 product candidate achieves positive clinical trial results, we may fail to obtain required regulatory approvals; |
|
● |
even if we obtain required regulatory approvals, the successful commercialization of our DM199 product candidate may fail to achieve market acceptance among physicians, patients, healthcare payors and the medical community; |
|
● |
if we fail to obtain coverage and adequate reimbursement for our DM199 product candidate, our revenue-generating ability will be diminished and there is no assurance that the anticipated market for our product will be sustained; |
|
● |
we face competition from other biotechnology and pharmaceutical companies and may face such competition sooner than expected if we do not qualify for data exclusivity as anticipated; and |
|
● |
we may be classified as a “passive foreign investment company,” which may have adverse U.S. federal income tax consequences for U.S. shareholders. |
6
Implications of Being an Emerging Growth Company
As a company with less than $1.07 billion of revenue during our last fiscal year, we are an emerging growth company as defined in the JOBS Act, and we may remain an emerging growth company for up to five years from the date of the first sale in this offering. However, if certain events occur prior to the end of such five-year period, including if we become a large accelerated filer, our annual gross revenue exceeds $1.07 billion, or we issue more than $1.0 billion of non-convertible debt in any three-year period, we will cease to be an emerging growth company prior to the end of such five-year period. For so long as we remain an emerging growth company, we are permitted and intend to rely on exemptions from certain disclosure and other requirements that are applicable to other public companies that are not emerging growth companies. In particular, in this prospectus, we have provided only two years of audited financial statements and have not included all of the executive compensation related information that would be required if we were not an emerging growth company. Accordingly, the information contained herein may be different than the information you receive from other public companies in which you hold equity interests. However, we have irrevocably elected not to avail ourselves of the extended transition period for complying with new or revised accounting standards, and, therefore, we are subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
Our Corporate Information
We are a corporation organized under CBCA. Our company was initially incorporated under the name Diabex Inc. pursuant to The Corporations Act (Manitoba) by articles of incorporation dated January 21, 2000. Our articles were amended (i) on February 26, 2001 to change our corporate name to DiaMedica Inc., (ii) on April 11, 2016 to continue the Company from The Corporations Act (Manitoba) to the CBCA, (iii) on December 28, 2016 to change our corporate name to DiaMedica Therapeutics Inc., (iv) on September 24, 2018 to permit us to hold shareholder meetings in the U.S. and to permit our directors, between annual meetings of our shareholders, to appoint one or more additional directors to serve until the next annual meeting of shareholders; provided, however, that the number of additional directors shall not at any time exceed one-third of the number of directors who held office at the expiration of the last meeting of shareholders, and (v) on November 15, 2018 to effect a 1-for-20 consolidation of our common shares.
Our registered office is located at 301-1665 Ellis Street, Kelowna, British Columbia, V1Y 2B3 and our principal executive office is located at our wholly owned subsidiary, DiaMedica USA Inc., located at 2 Carlson Parkway, Suite 260, Minneapolis, Minnesota, USA 55447. Our telephone number is 763-496-5454. Our internet website address is http://www.diamedica.com. Information contained on our website does not constitute part of this prospectus.
7
The Offering
The following summary contains basic information about this offering. The summary is not intended to be complete. You should read the full text and more specific details contained elsewhere in this prospectus.
|
Issuer |
DiaMedica Therapeutics Inc. |
|
Common shares offered by us |
shares |
|
Over-allotment option |
The underwriter has an option for a period of 30 days from the date of this prospectus to purchase up to additional common shares from us at the initial public offering price, less the underwriting discounts and commissions to cover over-allotments, if any. |
|
Common shares to be outstanding immediately after this offering(1) |
common shares (or common shares if the underwriter exercises its option to purchase additional shares in full). |
|
Use of proceeds |
We estimate that the net proceeds from this offering, after deducting the underwriting discounts and commissions and estimated offering expenses payable by us, will be approximately $ million, or approximately $ million if the underwriter exercises its over-allotment option to purchase additional common shares from us in full, based on an assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus. We intend to use the net proceeds from this offering to fund clinical development of DM199, to conduct research activities and for working capital and general corporate purposes. See “Use of Proceeds” for a more complete description of the intended use of proceeds from this offering. |
|
Dividend policy |
We do not expect to pay any dividends or other distributions on our common shares in the foreseeable future. We currently intend to retain future earnings. See “Dividend Policy.” |
|
Risk factors |
You should read the “Risk Factors” section of this prospectus and the other information in this prospectus for a discussion of factors to consider carefully before deciding to invest in our common shares. |
|
Proposed listing |
We have applied to have our common shares listed on The Nasdaq Capital Market. No assurance can be given that such listing will be approved. |
|
Proposed Nasdaq Capital Market symbol |
DMAC |
8
|
(1) |
The number of common shares to be outstanding after this offering is based on 7,839,176 common shares outstanding as of September 30, 2018 and excludes: |
|
● |
639,359 common shares issuable upon the exercise of stock options outstanding under the DiaMedica Therapeutics Inc. Stock Option Plan as of September 30, 2018, at a weighted-average exercise price of $7.87 per share; |
|
● |
21,183 common shares issuable upon the settlement of deferred share units outstanding under the DiaMedica Therapeutics Inc. Deferred Share Unit Plan as of September 30, 2018; |
|
● |
123,374 common shares reserved for future issuance under the DiaMedica Therapeutics Inc. Stock Option and DiaMedica Therapeutics Inc. Deferred Share Unit Plans as of September 30, 2018; |
|
● |
825,264 common shares issuable upon the exercise of outstanding warrants as of September 30, 2018, at a weighted average exercise price of $6.67 per share; and |
|
● |
common shares issuable upon the exercise of the warrant that will be issued to the underwriter in connection with this offering, with an exercise price equal to 120% of the initial public offering price per share. |
Except as otherwise indicated, all information in this prospectus reflects the consummation of the 1-for-20 share consolidation of our common shares effected on November 15, 2018 and assumes the following:
|
● |
no exercise of options or warrants described above after September 30, 2018; and |
|
● |
no exercise by the underwriter of its option to purchase additional common shares to cover over-allotments. |
9
SUMMARY CONSOLIDATED FINANCIAL DATA
The following tables set forth summary consolidated financial data of our company. The summary consolidated statements of operations and comprehensive loss data for the years ended December 31, 2017 and 2016 and the consolidated balance sheet data as of December 31, 2017 as set forth below are derived from our audited consolidated financial statements included elsewhere in this prospectus. The summary consolidated statements of operations and comprehensive loss data for the nine months ended September 30, 2018 and 2017 and the consolidated balance sheet data as of September 30, 2018 have been derived from our unaudited consolidated financial statements included elsewhere in this prospectus. These unaudited financial statements have been prepared on a basis consistent with our audited financial statements and, in the opinion of management, reflect all adjustments, consisting only of normal and recurring adjustments, necessary for a fair presentation of such financial data.
The information is only a summary and you should read it in conjunction with our audited consolidated financial statements, including the related notes, and other financial information and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this prospectus. Historical results are not necessarily indicative of the results for future periods.
|
Year Ended |
Nine Months |
|||||||||||||||
|
2016 |
2017 |
2017 |
2018 |
|||||||||||||
|
(unaudited) |
||||||||||||||||
|
(in thousands, exept share and per share data) |
||||||||||||||||
|
Consolidated Statements of Operations Data: |
||||||||||||||||
|
Operating revenues: |
||||||||||||||||
|
License revenue |
$ | - | $ | - | $ | - | $ | 500 | ||||||||
|
Operating expenses: |
||||||||||||||||
|
Research and development |
1,728 | 3,206 | 2,577 | 3,071 | ||||||||||||
|
General and administrative |
598 | 1,313 | 941 | 2,073 | ||||||||||||
|
Total operating expenses |
2,326 | 4,519 | 3,518 | 5,144 | ||||||||||||
|
Loss from operations |
(2,326 | ) | (4,519 | ) | (3,518 | ) | (4,644 | ) | ||||||||
|
Other (income) expense |
||||||||||||||||
|
Governmental assistance - research incentives |
- | (244 | ) | (244 | ) | (1,046 | ) | |||||||||
|
Other (income) expense |
82 | (6 | ) | 14 | 61 | |||||||||||
|
Change in fair value of warrant liability |
(188 | ) | (9 | ) | 208 | 39 | ||||||||||
|
Total other (income) expense |
(106 | ) | (259 | ) | (22 | ) | (946 | ) | ||||||||
|
Loss before income tax |
(2,220 | ) | (4,260 | ) | (3,496 | ) | (3,698 | ) | ||||||||
|
Income tax |
- | - | - | 74 | ||||||||||||
|
Net loss & comprehensive loss |
$ | (2,220 | ) | $ | (4,260 | ) | $ | (3,496 | ) | $ | (3,772 | ) | ||||
|
Loss per share, basic and diluted |
$ | (0.47 | ) | $ | (0.72 | ) | $ | (0.60 | ) | $ | (0.51 | ) | ||||
|
Weighted average number of shares outstanding: |
||||||||||||||||
|
Basic and diluted |
4,735,751 | 5,935,790 | 5,848,178 | 7,406,378 | ||||||||||||
|
Consolidated Balance Sheet Data: |
||||||||||||||||
|
As of December 31, |
As of |
|||||||||||||||
|
2016 |
2017 |
September 30, 2018 |
||||||||||||||
|
Actual |
Actual |
Actual |
As Adjusted |
|||||||||||||
|
(unaudited) |
||||||||||||||||
|
Cash |
$ | 1,736 | $ | 1,353 | $ | 3,898 | $ | |||||||||
|
Working capital |
1,092 | 491 | 3,748 | |||||||||||||
|
Total assets |
1,875 | 1,802 | 5,463 | |||||||||||||
|
Total current liabilities |
764 | 1,003 | 1,353 | |||||||||||||
|
Total stockholders' equity |
1,111 | 799 | 4,110 | |||||||||||||
10
RISK FACTORS
An investment in our common shares involves a high degree of risk and should be considered speculative. An investment in our common shares should only be undertaken by those persons who can afford the total loss of their investment. You should consider carefully the risks and uncertainties described below, as well as other information contained in this prospectus, including our consolidated financial statements and the related notes. The risks and uncertainties below are not the only ones we face. Additional risks and uncertainties not presently known to us or that we believe to be immaterial may also adversely affect our business. If any of the following risks occur, our business, financial condition, and results of operations could be seriously harmed and you could lose all or part of your investment. Further, if we fail to meet the expectations of the public market in any given period, the market price of our common shares could decline.
Risks Related to Our Financial Position and Need for Additional Capital
We have incurred substantial losses since our inception and expect to incur future losses and may never become profitable.
We are a clinical stage biopharmaceutical company focused on the development of novel recombinant proteins. Investment in biopharmaceutical product development is highly speculative because it entails substantial upfront capital expenditures and significant risk that a product candidate will fail to prove effective, gain regulatory approval or become commercially viable. We do not have any products approved by regulatory authorities and have not generated any revenues from collaboration and licensing agreements or product sales to date, and have incurred significant research, development and other expenses related to our ongoing operations and expect to continue to incur such expenses. As a result, we have not been profitable and have incurred significant operating losses in every reporting period since our inception. For the nine months ended September 30, 2018, we incurred a net loss of $3.8 million and for the years ended December 31, 2017 and 2016, we incurred a net loss of $4.3 million and $2.2 million, respectively. As of September 30, 2018, we had an accumulated deficit of $44.0 million. Our operating losses are expected to increase in the near term as we continue our product development efforts and are expected to continue until such time as any future product sales, royalty payments, licensing fees, and/or milestone payments are sufficient to generate revenues to fund our continuing operations. In addition, we expect to our operating expenses to increase compared to last year as a result of our U.S. public reporting company status. We are unable to predict the extent of any future losses or when we will become profitable, if ever. Even if we do achieve profitability, we may not be able to sustain or increase profitability on an ongoing basis.
We currently have no product revenue and will not be able to maintain our operations and research and development activities without additional funding.
To date, we have primarily relied on equity financing to fund our working capital requirements and drug development activities. A substantial amount of additional capital is needed to develop our product candidate, DM199, or any future product candidates to a point where they may be commercially sold. Our future operations are dependent upon our ability to generate product sales, negotiate collaboration or license agreements, obtain research grant funding, defer expenditures or other strategic alternatives, and/or secure additional funds. While we are striving to achieve these plans, there is no assurance these and other strategies will be achieved or that such sources of funds will be available or obtained on favorable terms or obtained at all. Our ability to continue as a going concern is dependent on our ability to continue obtaining sufficient funds to conduct our research and development (“R&D”) activities and to successfully commercialize our product candidates.
We will require additional funds to finance our operations, which may not be available to us on acceptable terms, or at all. As a result, we may not complete the development and commercialization of our current product candidate or develop new product candidates.
We require significant additional funds for further R&D activities, planned clinical trials and the regulatory approval process. We expect the net proceeds of this offering to be sufficient to allow us to complete our current Phase II Remedy trial in patients with acute ischemic stroke and a Phase 1b and Phase II study in patients with chronic kidney disease. We do not expect the net proceeds of this offering to be sufficient to fund, and we expect to require additional funding to complete, the development of DM199 through regulatory approval and commercialization, which we may seek through public or private equity or debt financings or through collaborations with other biotechnology companies or other sources. We may raise additional funds for these purposes through public or private equity or debt financing, or through collaborations with other biotechnology companies, or financing from other sources may be undertaken. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interests of our shareholders will be diluted. Debt financing, if available, may involve agreements that include conversion discounts or covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through government or other third-party funding, marketing and distribution arrangements or other collaborations, or strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us. It is possible that financing will not be available or, if available, may not be on favorable terms. The availability of financing will be affected by the results of scientific and clinical research; the ability to attain regulatory approvals; market acceptance of our product candidates; the state of the capital markets generally with particular reference to pharmaceutical, biotechnology, and medical companies; the status of strategic alliance agreements; and other relevant commercial considerations. If adequate funding is not available, we may be required to implement cost reduction strategies; delay, reduce, or eliminate one or more of our product development programs; relinquish significant rights to product candidates or obtain funds on less favorable terms than we would otherwise accept; and/or divest assets through a merger, sale, or liquidation of our company.
There is substantial doubt about our ability to continue as a going concern.
The report of our independent registered public accounting firm on our December 31, 2017 audited consolidated financial statements includes an explanatory paragraph referring to our ability to continue as a going concern. As of December 31, 2017 and September 30, 2018, we had cash balances of approximately $1.4 million and $3.9 million, respectively. In addition, we had outstanding accounts payable and accrued liabilities of $919,000 and $1.4 million as of December 31, 2017 and September 30, 2018, respectively. On March 29, 2018, we completed, in two tranches, a brokered and non-brokered private placement of 1,322,965 units at a price of $4.90 per unit for aggregate gross proceeds of approximately $6.3 million. Each unit consisted of one common share and one half of one common share purchase warrant. Each whole warrant entitles the holder to purchase one common share at a price of $7.00 at any time prior to March 19, 2020 and March 29, 2020 for the first and second tranches, respectively, subject to earlier expiration under certain conditions. Additional funding will be required to continue our R&D and other operating activities as we have not reached successful commercialization of our product candidates. These circumstances cast significant doubt as to our ability to continue as a going concern.
We are exposed to the financial risk related to the fluctuation of foreign exchange rates and the degrees of volatility of those rates.
We may be adversely affected by foreign currency fluctuations. To date, we have been primarily funded through issuances of equity and proceeds from the exercise of warrants and stock options, which are denominated both in Canadian and U.S. dollars. Currently, the majority of our expenditures are in U.S. dollars, however, significant costs are also incurred in Canadian dollars, British pounds, and Australian dollars; and, therefore, we are subject to foreign currency fluctuations which may, from time to time, impact our financial position and results of operations.
Risks Related to our Business and our Industry
We are an early stage company with no approved products and no revenue from commercialization of our products.
We are at an early stage of development of our product candidate, DM199, for the treatment of AIS and CKD. We have not completed the development of any product candidate and, accordingly, have not begun to commercialize, any product candidate or generate any product revenues from any product candidate. DM199 requires significant additional clinical testing and investment prior to seeking marketing approval. A commitment of substantial resources by ourselves and potential partners to continue to conduct clinical trials for DM199 will be required to meet applicable regulatory standards, obtain required regulatory approvals, and to successfully commercialize this product candidate. DM199 is not expected to be commercially available for several years, if at all.
Our prospects depend on the success of our product candidate, DM199, which is at an early stage of development, and we may not generate revenue for several years, if at all, from this product candidate or any future product candidates.
We are highly dependent on the success of DM199 and we may not be able to successfully obtain regulatory or marketing approval for, or successfully commercialize, this product candidate. To date, we have expended significant time, resources and effort on the development of DM199, including conducting preclinical and clinical trials, for the treatment of acute ischemic stroke and chronic kidney disease. Although we intend to study the use of DM199 to treat multiple diseases, we have no other product candidates in our current clinical development pipeline. Our ability to generate product revenues and to achieve commercial success in the near term will initially depend almost entirely on our ability to successfully develop, obtain regulatory approval for and then successfully commercialize DM199. Prior to commercialization of any potential product, significant additional investments will be necessary to complete the development of DM199 or any future product candidates. Preclinical and clinical trial work must be completed before DM199 or any future product candidate could be ready for use within the markets that we have identified. We may fail to develop any products, to obtain regulatory approvals, to enter clinical trials, or to commercialize any products. Competitors may develop alternative products and methodologies to diagnose and treat the disease indications we are pursuing, thus reducing our competitive advantages. We do not know whether any of our product development efforts will prove to be effective, meet applicable regulatory standards, obtain the requisite regulatory approvals, be capable of being manufactured at a reasonable cost, or successfully marketed. The product candidate we are currently developing is not expected to be commercially viable for several years. In addition, our product candidate may cause undesirable side effects. Results of early preclinical research may not be indicative of the results that will be obtained in later stages of preclinical or clinical research. If regulatory authorities do not approve our product candidate or any future product candidates or if we fail to maintain regulatory compliance, we would have limited ability to commercialize our product candidate or any future product candidates, and our business and results of operations would be harmed. If we do succeed in developing viable products from our product candidates, we will face many potential obstacles such as the need to develop or obtain manufacturing, marketing, and distribution capabilities.
We rely and will continue to rely on third parties to plan, conduct, and monitor our preclinical and clinical trials, and their failure to perform as required could cause substantial harm to our business.
We rely and will continue to rely on third parties to conduct a significant portion of our preclinical and clinical development activities. Preclinical activities include in vivo studies providing access to specific disease models, pharmacology and toxicology studies, and assay development. Clinical development activities include trial design, regulatory submissions, clinical patient recruitment, clinical trial monitoring, clinical data management and analysis, safety monitoring, and project management. If there is any dispute or disruption in our relationship with third parties, or if they are unable to provide quality services in a timely manner and at a feasible cost, our active development programs may face delays. Further, if any of these third parties fails to perform as we expect or if their work fails to meet regulatory requirements, our testing could be delayed, cancelled, or rendered ineffective.
We rely on a contract manufacturer over whom we have limited control. If we are subject to quality, cost, or delivery issues with the preclinical and clinical grade materials supplied by this or future contract manufacturers, our business operations could suffer significant harm.
We rely on a contract manufacturing organization (“CMO”) to manufacture our product candidate, DM199, for our preclinical studies and clinical trials. We rely on this CMO for manufacturing, filling, packaging, storing, and shipping of drug product in compliance with current good manufacturing practices (“GMP”) regulations applicable to our product candidate. The U.S. Food and Drug Administration (“FDA”) ensures the quality of drug products by carefully monitoring drug manufacturers’ compliance with “GMP” regulations. The “GMP” regulations for drugs contain minimum requirements for the methods, facilities, and controls used in manufacturing, processing, and packing of a drug product.
There can be no assurances that this CMO will be able to meet our timetable and requirements. If we are unable to arrange for alternative third-party manufacturing sources on commercially reasonable terms or in a timely manner, we may be delayed in the development of DM199 and any future product candidates. Further, CMOs must operate in compliance with GMP regulations and failure to do so could result in, among other things, the disruption of product supplies. Our dependence upon this CMO and any future third parties for the manufacture of our product candidates may adversely affect our ability to develop our product candidates on a timely and competitive basis and, if we are able to commercialize our product candidates, may adversely affect our profit margins.
If clinical trials of our product candidates fail to demonstrate safety and efficacy to the satisfaction of regulatory authorities or do not otherwise produce positive results, we would incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates.
Before obtaining marketing approval from regulatory authorities for the sale of our product candidates, we must conduct preclinical studies in animals and extensive clinical trials in humans to demonstrate the safety and efficacy of our product candidates. Clinical testing is expensive and difficult to design and implement, can take many years to complete, and has uncertain outcomes. The outcome of preclinical studies and early clinical trials may not predict the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced clinical trials due to lack of efficacy or unacceptable safety profiles, notwithstanding promising results in earlier trials. We do not know whether the clinical trials we may conduct will demonstrate adequate efficacy and safety to result in regulatory approval to market any of our product candidates in any jurisdiction. A product candidate may fail for safety or efficacy reasons at any stage of the testing process. A major risk we face is the possibility that neither our current or future product candidates will successfully gain market approval from the FDA or other regulatory authorities, resulting in us being unable to derive any commercial revenue from them after investing significant amounts of capital in multiple stages of preclinical and clinical testing.
If we experience delays in clinical testing, we will be delayed in commercializing our product candidates, and our business may be substantially harmed.
We cannot predict whether any clinical trials will begin as planned, will need to be restructured, or will be completed on schedule, or at all. Our product development costs will increase if we experience delays in clinical testing. Significant clinical trial delays could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before us, which would impair our ability to successfully commercialize our product candidates and may harm our financial condition, results of operations and prospects. The commencement and completion of clinical trials for our product candidates may be delayed for a number of reasons, including delays related, but not limited, to:
|
● |
failure by regulatory authorities to grant permission to proceed or placing the clinical trial on hold; |
|
● |
patients failing to enroll or remain in our trials at the rate we expect; |
|
● |
suspension or termination of clinical trials by regulators for many reasons, including concerns about patient safety or failure of our contract manufacturers to comply with GMP requirements; |
|
● |
any changes to our manufacturing process that may be necessary or desired; |
|
● |
delays or failure to obtain clinical supply from contract manufacturers of our product candidates necessary to conduct clinical trials; |
|
● |
product candidates demonstrating a lack of safety or efficacy during clinical trials; |
|
● |
patients choosing an alternative treatment for the indications for which we are developing any of our product candidates or participating in competing clinical trials; |
|
● |
patients failing to complete clinical trials due to dissatisfaction with the treatment, side effects, or other reasons; |
|
● |
reports of clinical testing on similar technologies and products raising safety and/or efficacy concerns; |
|
● |
competing clinical trials and scheduling conflicts with participating clinicians; |
|
● |
clinical investigators not performing our clinical trials on their anticipated schedule, dropping out of a trial, or employing methods not consistent with the clinical trial protocol, regulatory requirements or other third parties not performing data collection and analysis in a timely or accurate manner; |
|
● |
failure of our contract research organizations (“CROs”) to satisfy their contractual duties or meet expected deadlines; |
|
● |
inspections of clinical trial sites by regulatory authorities, Institutional Review Boards (“IRBs”) or ethics committees finding regulatory violations that require us to undertake corrective action, resulting in suspension or termination of one or more sites or the imposition of a clinical hold on the entire study; |
|
● |
one or more IRBs or ethics committees rejecting, suspending or terminating the study at an investigational site, precluding enrollment of additional subjects, or withdrawing its approval of the trial; or |
|
● |
failure to reach agreement on acceptable terms with prospective clinical trial sites. |
Our product development costs will increase if we experience delays in testing or approval or if we need to perform more or larger clinical trials than planned. Additionally, changes in regulatory requirements and policies may occur, and we may need to amend study protocols to reflect these changes. Amendments may require us to resubmit our study protocols to regulatory authorities or IRBs or ethics committees for re-examination, which may impact the cost, timing or successful completion of that trial. Delays or increased product development costs may have a material adverse effect on our business, financial condition, and prospects.
Even if we complete the necessary preclinical studies and clinical trials, the regulatory approval process is expensive, time-consuming and uncertain and may prevent us or any future collaborators from obtaining approvals for the commercialization of some or all of our product candidates. As a result, we cannot predict when or if, and in which territories, we, or any future collaborators, will obtain marketing approval to commercialize a product candidate.
The process of obtaining marketing approvals, both in the United States and abroad, is expensive and may take many years, if approval is obtained at all, and can vary substantially based upon a variety of factors, including the type, complexity and novelty of the product candidates involved.
Our current product candidate and the activities associated with its development and commercialization, including design, research, testing, manufacture, safety, efficacy, quality control, recordkeeping, labeling, packaging, storage, approval, advertising, promotion, sale, distribution, import, export, and reporting of safety and other post-market information, are subject to comprehensive regulation by the FDA, the European Medicines Agency (“EMA”) and other foreign regulatory agencies. Failure to obtain marketing approval for a product candidate will prevent us from commercializing the product candidate. We have only limited experience in filing and supporting the applications necessary to gain marketing approvals and expect to rely on third-parties to assist us in this process. Securing marketing approval requires the submission of extensive preclinical and clinical data and supporting information to regulatory authorities for each therapeutic indication to establish the product candidate's safety and efficacy. Securing marketing approval also requires the submission of information about the product manufacturing process to, and inspection of manufacturing facilities by, the regulatory authorities. The FDA, EMA or other regulatory authorities may determine that our product candidates may not be effective, may be only moderately effective or may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude our obtaining marketing approval or prevent or limit commercial use. As a result, any marketing approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render the approved product not commercially viable.
In addition, changes in marketing approval policies during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for each submitted product application, may cause delays in the approval or rejection of an application. Regulatory authorities have substantial discretion in the approval process and may refuse to accept any application or may decide that our data is insufficient for approval and require additional preclinical, clinical or other studies. In addition, varying interpretations of the data obtained from preclinical and clinical testing could delay, limit or prevent marketing approval of a product candidate.
We are in litigation with Pharmaceutical Research Associates Group B.V., a contract research organization, seeking to compel them to comply with the terms of a clinical trial research agreement and their failure to perform as required could adversely affect our ability to obtain regulatory approval for DM199.
In March 2013, we entered into a clinical research agreement with Pharmaceutical Research Associates Group B.V. (“PRA”) to perform a double-blinded, placebo-controlled, single-dose and multiple-dose study to evaluate the safety, tolerability, pharmacokinetics, pharmacodynamics and proof of concept of DM199 in healthy subjects and in patients with Type 2 diabetes mellitus. In one arm of this study, we enrolled 36 patients with Type 2 diabetes who were treated with two SC dose levels of DM199 over a 28-day period. This study achieved its primary endpoint and demonstrated that DM199 was well tolerated. The secondary endpoints for this study, however, were not met. We believe there were significant execution errors in Part D of the study that were caused by protocol deviations occurring at the clinical trial site that were unable to be reconciled. We believe these included dosing errors and sample mix-ups. These errors undermined our ability to interpret the secondary endpoints. To date, we have been unable to obtain the complete study records from PRA for the arm of the study which included 36 patients with Type 2 diabetes and was intended to measure primary endpoints (safety, tolerability) and secondary endpoints (blood glucose concentration, insulin levels, glucose tolerance test and a variety of experimental biomarkers). Without these records and given our inability to reconcile the protocol deviations, we have been unable to generate a final study report. Due in part to these confounded secondary endpoints, we are not currently continuing the clinical study of DM199 for Type 2 diabetes. We believe that the consistently positive safety and tolerability demonstrated in our studies to date will allow us to pursue approval for the clinical study of DM199 in the treatment of CKD patients in the United States; however, the lack of a final study report may delay or prevent our ability to obtain the acceptance of an Investigational New Drug (“IND”) which would delay or prevent us from conducting clinical development or obtaining approval in the United States. We have initiated litigation with PRA to compel them to comply with the terms of the clinical research agreement, including providing full study records, and to recover damages. Litigation distracts the attention of our management from our business, is expensive and the outcome is uncertain.
We may not be able to obtain FDA acceptance of INDs to commence clinical trials in the United States on the timelines we expect, and even if we are able to, the FDA may not permit us to proceed in a timely manner, or at all.
Prior to commencing clinical trials in the United States for our current or any future product candidates, we will likely be required to have an accepted IND for each product candidate and for each targeted indication. We have not yet filed an IND to initiate a clinical trial for DM199 in the United States. However, submission of an IND may not result in the FDA allowing further clinical trials to begin and, once begun, issues may arise that will require us to suspend or terminate such clinical trials. Additionally, even if relevant regulatory authorities agree with the design and implementation of the clinical trials set forth in an IND, these regulatory authorities may change their requirements in the future. Failure to submit or have effective INDs and commence clinical programs will significantly limit our opportunity to generate revenue.
If we have difficulty enrolling patients in clinical trials, the completion of the trials may be delayed or not completed at all.
As DM199 and any future product candidates advance from preclinical testing to clinical testing, and then through progressively larger and more complex clinical trials, we will need to enroll an increasing number of patients that meet our eligibility criteria. There is significant competition for recruiting patients in clinical trials, and we may be unable to enroll the patients we need to complete clinical trials on a timely basis or at all. The factors that affect our ability to enroll patients are largely uncontrollable and include, but are not limited to, the following:
|
● |
size and nature of the patient population; |
|
● |
eligibility and exclusion criteria for the trial; |
|
● |
design of the study protocol; |
|
● |
competition with other companies for clinical sites or patients; |
|
● |
the perceived risks and benefits of the product candidate under study; |
|
● |
the patient referral practices of physicians; and |
|
● |
the number, availability, location, and accessibility of clinical trial sites. |
We may not be able to reproduce the results of previously conducted clinical studies and/or comparisons to other forms of KLK1, including Kailikang®, thereby displacing other forms of KLK1, including Kailikang®.
While there have been numerous studies demonstrating the efficacy of Kailikang®, we rely on the scientific and clinical knowledge and experience of other biotechnology and pharmaceutical companies and organizations in conducting those clinical studies. No assurance can be given that in our clinical trials involving DM199 we will be able to reproduce results of previously conducted studies or displace other forms of KLK1 in the market.
Negative results from clinical trials or studies of others and adverse safety events involving the targets of our product candidates may have an adverse impact on our future commercialization efforts.
From time to time, studies or clinical trials on various aspects of biopharmaceutical products are conducted by academic researchers, competitors, or others. The results of these studies or trials, when published, may have a significant effect on the market for the biopharmaceutical product that is the subject of the study. The publication of negative results of studies or clinical trials or adverse safety events related to our product candidates, or the therapeutic areas in which our product candidates compete, could adversely affect our share price and our ability to finance future development of our product candidates, and our business and financial results could be materially and adversely affected.
We may be required to suspend, repeat or terminate our clinical trials if they are not conducted in accordance with regulatory requirements, the results are negative or inconclusive, or the trials are not well designed.
Clinical trials must be conducted in accordance with the FDA’s current Good Clinical Practices requirements, or cGCPs, or analogous requirements of applicable foreign regulatory authorities. Clinical trials are subject to oversight by the FDA, other foreign governmental agencies, and IRBs or ethics committees at the study sites where the clinical trials are conducted. In addition, clinical trials must be conducted with product candidates produced in accordance with applicable current Good Manufacturing Practices. Clinical trials may be suspended by us or by the FDA, other foreign regulatory authorities, or by an IRB or ethic committee with respect to a particular clinical trial site, for various reasons, including:
|
● |
deficiencies in the conduct of the clinical trials, including failure to conduct the clinical trial in accordance with regulatory requirements or study protocols; |
|
● |
deficiencies in the clinical trial operations or trial sites; |
|
● |
unforeseen adverse side effects or the emergence of undue risks to study subjects; |
|
● |
deficiencies in the trial design necessary to demonstrate efficacy; |
|
● |
the product candidate may not appear to offer benefits over current therapies; or |
|
● |
the quality or stability of the product candidate may fall below acceptable standards. |
The design and implementation of clinical trials is a complex process. As a Company, we have limited experience designing and implementing clinical trials, and failure to adequately design a trial, or incorrect assumptions about the design of the trial, could adversely affect the ability to initiate the trial, enroll patients, complete the trial, or obtain regulatory approval on the basis of the trial results, as well as lead to increased or unexpected costs. We may not successfully or cost-effectively design and implement clinical trials that achieve our desired clinical endpoints efficiently, or at all. A clinical trial that is not well designed may delay or even prevent initiation of the trial, can lead to increased difficulty in enrolling patients, may make it more difficult to obtain regulatory approval for the product candidate on the basis of the study results, or, even if a product candidate is approved, could make it more difficult to commercialize the product successfully or obtain reimbursement from third party payors. Additionally, a trial that is not well-designed could be inefficient or more expensive than it otherwise would have been, or we may incorrectly estimate the costs to implement the clinical trial, which could lead to a shortfall in funding.
Regulatory approval processes are lengthy, expensive, and inherently unpredictable. Our inability to obtain regulatory approval for our product candidates would substantially harm our business.
Our shareholders and other investors should be aware of the risks, problems, delays, expenses, and difficulties which we may encounter in light of the extensive regulatory environment within which our business is carried out. Numerous statutes and regulations govern the preclinical and clinical development, manufacture and sale, and post-marketing responsibilities for non-therapeutic and human therapeutic products in the United States, European Union, Canada, Australia and other countries that are the intended markets for our current and future product candidates. Such legislation and regulation governs the approval of manufacturing facilities, the testing procedures, and controlled research that must be carried out, and the preclinical and clinical data that must be collected prior to marketing approval. Our R&D efforts, as well as any future clinical trials, and the manufacturing and marketing of any products we may develop, will be subject to and restricted by such extensive regulation.
The process of obtaining necessary regulatory approvals is lengthy, expensive, and uncertain. We may fail to obtain the necessary approvals to commence or continue clinical testing or to manufacture or market our potential products in reasonable time frames, if at all. In addition, governmental authorities in the United States or other countries may enact regulatory reforms or restrictions on the development of new therapies that could adversely affect the regulatory environment in which we operate or the development of any products we may develop.
Completing clinical testing and obtaining required approvals is expected to take several years and to require the expenditure of substantial resources. There can be no assurance that clinical trials will be completed successfully within any specified period of time, if at all. Furthermore, clinical trials may be delayed or suspended at any time by us or by the various regulatory authorities if it is determined at any time that the subjects or patients are being exposed to unacceptable risks.
Any failure or delay in obtaining regulatory approvals would adversely affect our ability to utilize our technology and would therefore adversely affect our operations. Furthermore, no assurance can be given that our current or future product candidates will prove to be safe and effective in clinical trials or that they will receive the requisite regulatory approval. Moreover, any regulatory approval of a drug which is eventually obtained may be granted with specific limitations on the indicated uses for which that drug may be marketed. Furthermore, product approvals may be withdrawn if problems occur following initial marketing or if compliance with regulatory standards is not maintained.
Any product candidate for which we obtain marketing approval could be subject to post-marketing restrictions or recall or withdrawal from the market, and we may be subject to penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with our product candidates, when and if any of them are approved.
The FDA and other federal and state agencies, including the U.S. Department of Justice (“DOJ”), closely regulate compliance with all requirements governing prescription drug products, including requirements pertaining to marketing and promotion of drugs in accordance with the provisions of the approved labeling and manufacturing of products in accordance with GMP requirements. The FDA and DOJ impose stringent restrictions on manufacturers' communications regarding off-label use and if we do not market our products for their approved indications, we may be subject to enforcement action for off-label marketing. Violations of such requirements may lead to investigations alleging violations of the Food, Drug and Cosmetic Act and other statutes, including the False Claims Act and other federal and state health care fraud and abuse laws as well as state consumer protection laws.
Our failure to comply with all regulatory requirements, and later discovery of previously unknown adverse events or other problems with our products, manufacturers or manufacturing processes, may yield various results, including:
|
● |
litigation involving patients taking our products; |
|
● |
restrictions on such products, manufacturers or manufacturing processes; |
|
● |
restrictions on the labeling or marketing of a product; |
|
● |
restrictions on product distribution or use; |
|
● |
requirements to conduct post-marketing studies or clinical trials; |
|
● |
warning or untitled letters; |
|
● |
withdrawal of the products from the market; |
|
● |
refusal to approve pending applications or supplements to approved applications that we submit; |
|
● |
recall of products; |
|
● |
fines, restitution or disgorgement of profits or revenues; |
|
● |
suspension or withdrawal of marketing approvals; |
|
● |
damage to relationships with any potential collaborators; |
|
● |
unfavorable press coverage and damage to our reputation; |
|
● |
refusal to permit the import or export of our products; |
|
● |
product seizure; or |
|
● |
injunctions or the imposition of civil or criminal penalties. |
Non-compliance by us or any future collaborator with regulatory requirements regarding safety monitoring or pharmacovigilance, and with requirements related to the development of products for the pediatric population, can also result in significant financial penalties. Similarly, failure to comply with regulatory requirements regarding the protection of personal information can also lead to significant penalties and sanctions.
Non-compliance with European Union requirements regarding safety monitoring or pharmacovigilance, and with requirements related to the development of products for the pediatric population, also can result in significant financial penalties. Similarly, failure to comply with the European Union’s requirements regarding the protection of personal information can also lead to significant penalties and sanctions.
We may not achieve our publicly announced milestones according to schedule, or at all.
From time to time, we may announce the timing of certain events we expect to occur, such as the anticipated timing of results from our clinical trials. These statements are forward-looking and are based on the best estimates of management at the time relating to the occurrence of such events. However, the actual timing of such events may differ significantly from what has been publicly disclosed. The timing of events such as the initiation or completion of a clinical trial, filing of an application to obtain regulatory approval, or an announcement of additional clinical trials for a product candidate may ultimately vary from what is publicly disclosed. These variations in timing may occur as a result of different events, including the nature of the results obtained during a clinical trial or during a research phase, problems with a CMO or CRO or any other event having the effect of delaying the publicly announced timeline. We undertake no obligation to update or revise any forward-looking information, whether as a result of new information, future events or otherwise, except as otherwise required by law. Any variation in the timing of previously announced milestones could have a material adverse effect on our business plan, financial condition or operating results, and the trading price of our common shares.
Future development collaborations may be important to us. If we are unable to enter into or maintain these collaborations, or if these collaborations are not successful, our business could be adversely affected.
We may in the future determine to seek to collaborate with pharmaceutical and biotechnology companies for development or commercialization of our current or future product candidates. We face significant competition in seeking appropriate collaborators. Our ability to reach a definitive agreement for any collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. If we are unable to reach agreements with suitable collaborators on a timely basis, on acceptable terms, or at all, we may have to curtail the development of a product candidate, reduce or delay its development program or one or more of our other development programs, delay its potential development schedule or reduce the scope of research activities, or increase our expenditures and undertake discovery, nonclinical or clinical development activities at our own expense. If we fail to enter into collaborations and do not have sufficient funds or expertise to undertake the necessary development activities, we may not be able to continue or further develop our current or future product candidates and our business may be materially and adversely affected.
Future collaborations we may enter into may involve the following risks:
|
● |
collaborators may have significant discretion in determining the efforts and resources that they will apply to these collaborations; |
|
● |
collaborators may not perform their obligations as expected; |
|
● |
changes in the collaborators’ strategic focus or available funding, or external factors, such as an acquisition, may divert resources or create competing priorities; |
|
● |
collaborators may delay discovery, nonclinical or clinical development, provide insufficient funding for product development of targets selected by us, stop or abandon discovery, nonclinical or clinical development for a product candidate, or repeat or conduct new discovery, and nonclinical and clinical development for a product candidate; |
|
● |
collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our products or product candidates if the collaborators believe that competitive products are more likely to be successfully developed than our products; |
|
● |
product candidates discovered in collaboration with us may be viewed by our collaborators as competitive with their own product candidates or products, which may cause collaborators to cease to devote resources to the development of our product candidates; |
|
● |
disagreements with collaborators, including disagreements over proprietary rights, contract interpretation or the preferred course of development, might cause delays or termination of the discovery, preclinical or clinical development or commercialization of product candidates, might lead to additional responsibilities for us with respect to product candidates, or might result in litigation or arbitration, any of which would be time-consuming and expensive; |
|
● |
collaborators may not properly maintain or defend our intellectual property rights or intellectual property rights licensed to us or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation; |
|
● |
collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability; and |
|
● |
collaborations may be terminated for the convenience of the collaborator and, if terminated, we could be required to raise additional capital to pursue further development or commercialization of the applicable product candidates. |
Additionally, subject to its contractual obligations to us, if a collaborator is involved in a business combination, the collaborator might deemphasize or terminate the development of any of our product candidates. If one of our collaborators terminates its agreement with us, we may find it more difficult to attract new collaborators and our perception in the business and financial communities could be adversely affected.
If our collaborations do not result in the successful development of products or product candidates, product candidates could be delayed and we may need additional resources to develop product candidates. All of the risks relating to product development, regulatory approval and commercialization described in this prospectus also apply to the activities of our collaborators.
We recently entered into a license and collaboration agreement with Ahon Pharma which allows the licensee to have exclusive rights to develop and commercialize DM199 for AIS in mainland China, Taiwan, Hong Kong S.A.R. and Macau S.A.R. in exchange for an upfront cash payment, potential future milestone payments and sales royalties. As a result, we are dependent upon this licensee for such development and commercialization and are not guaranteed of receipt of the potential future milestone payments and sales royalties.
We recently entered into a license and collaboration agreement with Ahon Pharma, a subsidiary of Fosun Pharma, which grants Ahon Pharma exclusive rights to develop and commercialize DM199 for AIS in mainland China, Taiwan, Hong Kong S.A.R. and Macau S.A.R. Under the terms of the agreement, we are entitled to receive an upfront payment of $5.0 million, consisting of $500,000 on signing and $4.5 million upon regulatory clearance to initiate a clinical trial in China. We also have the potential to receive an additional $27.5 million in development and sales related milestones and up to approximately 10% royalties on net sales of DM199 in the licensed territories. All development, regulatory, sales, marketing, and commercial activities and associated costs in the licensed territories will be the sole responsibility of Ahon Pharma. As a result, we are dependent upon Ahon Pharma for such development and commercialization. There can be no assurance that we will receive the potential future milestone payments and sales royalties. This agreement may be terminated at any time by Ahon Pharma by providing 120 days written notice.
The successful commercialization of our current or future product candidates, if approved, will depend on achieving market acceptance and we may not be able to gain sufficient acceptance to generate significant revenue.
Even if our product candidates are successfully developed and receive regulatory approval, they may not gain market acceptance among physicians, patients, healthcare payors such as private insurers or governments and other funding parties, and the medical community. The degree of market acceptance for any products we develop will depend on a number of factors, including:
|
● |
demonstration of the clinical efficacy and safety; |
|
● |
the prevalence and severity of any adverse side effects; |
|
● |
limitations or warnings contained in the product’s approved labeling; |
|
● |
cost-effectiveness and availability of acceptable pricing; |
|
● |
competitive product profile versus alternative treatment methods and the superiority of alternative treatment or therapeutics; |
|
● |
the effectiveness of marketing and distribution methods and support for the products; and |
|
● |
coverage and reimbursement policies of government and third-party payors to the extent that our products could receive regulatory approval but not be approved for coverage by or receive adequate reimbursement from government and quasi-government agencies or other third-party payors. |
Disease indications may be small subsets of a disease that could be parsed into smaller and smaller indications as different subsets of diseases are defined. This increasingly fine characterization of diseases could have negative consequences; including creating an approved indication that is so small as not to have a viable market for us. If future technology allows characterization of a disease in a way that is different from the characterization used for large pivotal studies, it may make those studies invalid or reduce their usefulness, and may require repeating all or a portion of the studies. Future technology may supply better prognostic ability which could reduce the portion of patients projected to need a new therapy. Even after being cleared by regulatory authorities, a product may later be shown to be unsafe or not to have its purported effect, thereby preventing its widespread use or requiring withdrawal from the market.
If we fail to obtain coverage and adequate reimbursement for our products, our revenue-generating ability will be diminished and there is no assurance that the anticipated market for our products will be sustained.
We believe that there may be many different applications for products successfully derived from our technologies and that the anticipated market for products under development will continue to expand. However, due to competition from existing or new products and the yet-to-be established commercial viability of our products, no assurance can be given that these beliefs will prove to be correct. Physicians, patients, formularies, payors or the medical community in general may not accept or utilize any products that we may develop. Other drugs may be approved during our clinical testing which could change the accepted treatments for the disease targeted and make our product candidates obsolete.
Our ability to commercialize our future products, if any, successfully will depend, in part, on the extent to which coverage and adequate reimbursement for such products and related treatments will be available from governmental health payor programs at the federal and state levels, including Medicare and Medicaid, private health insurers, managed care plans and other organizations. No assurance can be given that third-party coverage and adequate reimbursement will be available that will allow us to maintain price levels sufficient for the realization of an appropriate return on our investment in product development. Coverage and adequate reimbursement from governmental healthcare programs, such as Medicare and Medicaid, and private health insurers, managed care plans and other organizations is critical to new product acceptance. There is no uniform coverage and reimbursement policy among third-party payors in the United States; however, private third-party payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates. Additionally, coverage decisions may depend upon clinical and economic standards that disfavor new drug products when more established or lower cost therapeutic alternatives are already available or subsequently become available. Even if we obtain coverage for our product candidates, the resulting reimbursement payment rates might not be adequate or may require co-payments that patients find unacceptably high. In addition, healthcare reform and controls on healthcare spending may limit the price we charge for any products and the amounts thereof that we can sell. Patients are unlikely to use our product candidates unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of our product candidates.
Outside of the United States, the successful commercialization of our products will depend largely on obtaining and maintaining government coverage, because in many countries patients are unlikely to use prescription drugs that are not covered by their government healthcare programs. Negotiating coverage and reimbursement with governmental authorities can delay commercialization by 12 months or more. Coverage and reimbursement policies may adversely affect our ability to sell our products on a profitable basis. In many international markets, governments control the prices of prescription pharmaceuticals, including through the implementation of reference pricing, price cuts, rebates, revenue-related taxes and profit control, and we expect prices of prescription pharmaceuticals to decline over the life of the product or as volumes increase.
We will not be able to successfully commercialize our current or future product candidates without establishing sales and marketing capabilities internally or through collaborators.
We currently have no sales and marketing staff. We may not be able to find suitable sales and marketing staff and collaborators for our product candidates. We have no prior experience in the marketing, sale and distribution of pharmaceutical products and there are significant risks involved in building and managing a sales organization, including our ability to hire, retain and incentivize qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel, and effectively manage a geographically dispersed sales and marketing team. Any collaborators may not be adequate or successful or could terminate or materially reduce the effort they direct to our products. The development of a marketing and sales capability will require significant expenditures, management resources and time. The cost of establishing such a sales force may exceed any potential product revenue, or our marketing and sales efforts may be unsuccessful. If we are unable to develop an internal marketing and sales capability in a timely fashion, or at all, or if we are unable to enter into a marketing and sales arrangement with a third party on acceptable terms, we may be unable to successfully develop and seek regulatory approval for our product candidates and/or effectively market and sell approved products, if any.
We face competition from other biotechnology and pharmaceutical companies and our financial condition and operations will suffer if we fail to effectively compete.
Technological competition is intense in the industry in which we operate. Competition comes from pharmaceutical companies, biotechnology companies, and universities, as well as companies that offer non-pharmaceutical solutions in the markets we may attempt to address with our products. Many of our competitors have substantially greater financial and technical resources; more extensive R&D capabilities; and greater marketing, distribution, production, and human resources than we do. Moreover, competitors may develop products more quickly than us and may obtain regulatory approval for such products more rapidly than we do. Products and processes which are more effective than those that we intend to develop may be developed by our competitors. R&D by others may render our product candidates non-competitive or obsolete.
Our product candidates may face competition sooner than expected.
We believe that DM199 could qualify for 12 years of data exclusivity in the United States under the Biologics Price Competition and Innovation Act of 2009 (“BPCIA”), which was enacted as part of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (collectively, the “ACA”). Under the BPCIA, an application for a biosimilar product, or BLA, cannot be submitted to the FDA until four years, or if approved by the FDA, until 12 years, after the original brand product identified as the reference product is approved under a BLA. The BPCIA provides an abbreviated pathway for the approval of biosimilar and interchangeable biological products. The new abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on its similarity to an existing brand product. The new law is complex and is only beginning to be interpreted and implemented by the FDA. While it is uncertain when any such processes may be fully adopted by the FDA, any such processes could have a material adverse effect on the future commercial prospects for any of our product candidates that are biologics. There is also a risk that the U.S. Congress could repeal or amend the BPCIA to shorten this exclusivity period, potentially creating the opportunity for biosimilar competition sooner than anticipated after the expiration of our patent protection. Moreover, the extent to which a biosimilar, once approved, will be substituted for any reference product in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing.
Even if, as we expect, our current or future product candidates are considered to be reference products eligible for 12 years of exclusivity under the BPCIA, another company could market competing products if the FDA approves a full BLA for such product containing the sponsor’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of the products. Moreover, an amendment or repeal of the BPCIA could result in a shorter exclusivity period for our product candidates, which could have a material adverse effect on our business.
Our relationships with customers and third-party payers will be subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, program exclusion, contractual damages, reputational harm and diminished profits and future earnings.
Healthcare providers, physicians and third-party payers will likely play a primary role in the recommendation and prescription of any product candidates for which we receive marketing approval. Our future arrangements with third-party payers and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute the products for which we receive marketing approval. Currently, restrictions under applicable federal and state healthcare laws and regulations include the following:
|
● |
the federal healthcare anti-kickback statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under federal and state healthcare programs such as Medicare and Medicaid. This statute may apply to our marketing practices, educational programs, pricing policies and relationships with healthcare providers. A person or entity does not need to have actual knowledge of this statute or specific intent to violate it to have committed a violation; |
|
● |
the federal False Claims Act imposes civil penalties, including civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. The government also may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the false claims statutes. Pharmaceutical and other healthcare companies have been prosecuted under these laws for, among other things, allegedly inflating drug prices they report to pricing services, which in turn were used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, certain marketing practices, including off-label promotion, may also violate false claims laws; |
|
● |
the federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), as amended by the Health Information Technology for Economic and Clinical Health Act, imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program and also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
|
● |
the federal false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services; |
|
● |
the federal transparency requirements under the ACA, require manufacturers of covered drugs, devices, biologics and medical supplies to report to the Department of Health and Human Services information related to physician payments and other transfers of value and physician ownership and investment interests; |
|
● |
analogous state laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payers, including private insurers, and some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring drug manufacturers to report information related to payments to physicians and other healthcare providers or marketing expenditures; |
|
● |
the distribution of prescription pharmaceutical products is subject to the Prescription Drug Marketing Act (“PDMA”) and its implementation regulations, as well as the Drug Supply Chain Security Act (“DSCSA”), which regulates the distribution of and tracing of prescription drugs and prescription drug samples at the federal level, and sets minimum standards for the regulation of drug distributors by the states. The PDMA, its implementing regulations and state laws limit the distribution of prescription pharmaceutical product samples, and the DSCSA imposes requirements to ensure accountability in distribution and to identify and remove counterfeit and other illegitimate products from the market; and |
|
● |
the U.S. Foreign Corrupt Practices Act of 1977, as amended (“FCPA”), the U.S. domestic bribery statute contained in 18 U.S.C. §201, the U.S. Travel Act. |
Some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government and may require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures. State and foreign laws also govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
The risk of our being found in violation of these laws and regulations is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. We are unable to predict what additional federal or state legislation or regulatory initiatives may be enacted in the future regarding our business or the healthcare industry in general, or what effect such legislation or regulations may have on us. Federal or state governments may impose additional restrictions or adopt interpretations of existing laws that could have a material adverse effect on us.
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any of the physicians or other providers or entities with whom we expect to do business are found to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Laws, restrictions, and other regulatory measures are also imposed by anti-kickback, fraud and abuse, and other healthcare laws and regulations in international jurisdictions and in those jurisdictions we face the same issues as in the United State regarding exposure to criminal sanctions, civil penalties, program exclusion, contractual damages, reputational harm, and diminished profits and future earnings.
We heavily rely on the capabilities and experience of our key executives and scientists and the loss of any of them could affect our ability to develop our products.
We depend on our management personnel. We also depend on our scientific and clinical collaborators and advisors, all of whom have outside commitments that may limit their availability to us. In addition, we believe that our future success will depend in large part upon our ability to attract and retain highly skilled scientific, managerial, medical, clinical, and regulatory personnel, particularly as we expand our activities and seek regulatory approvals for clinical trials. We enter into agreements with scientific and clinical collaborators and advisors, key opinion leaders, and academic partners in the ordinary course of our business. We also enter into agreements with physicians and institutions who will recruit patients into our clinical trials on our behalf in the ordinary course of our business. Notwithstanding these arrangements, we face significant competition for these types of personnel from other companies, research and academic institutions, government entities and other organizations. We cannot predict our success in hiring or retaining the personnel we require for continued growth. The loss of the services of any of our executive officers or other key personnel could potentially harm our business, operating results, or financial condition.
We will likely need to expand our operations and increase the size of our company, and we may experience difficulties in managing growth.
As we advance DM199 and any future product candidates through preclinical testing and clinical studies, and develop our current or future product candidates, we will need to increase our product development, scientific, regulatory and compliance and administrative headcount to manage these programs. In addition, to meet our obligations as a public company, we will need to increase our general and administrative capabilities. Our management, personnel and systems currently in place may not be adequate to support this future growth. Our need to effectively manage our operations, growth and various projects requires that we:
|
● |
successfully attract and recruit new employees with the expertise and experience we will require; |
|
● |
manage our clinical programs effectively, which we anticipate being conducted at numerous clinical sites; |
|
● |
develop a marketing, distribution and sales infrastructure if we seek to market our products directly; and |
|
● |
continue to improve our operational, manufacturing, quality assurance, financial and management controls, reporting systems and procedures. |
If we are unable to successfully manage this growth and increased complexity of operations, our business may be adversely affected.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could have a material adverse effect on our business.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include failures to comply with FDA regulations, provide accurate information to the FDA, comply with manufacturing and reporting standards we have established, comply with federal and state health-care fraud and abuse laws and regulations, report financial information or data accurately, or disclose unauthorized activities to us. In particular, sales, marketing, and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing, and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs, and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a substantial impact on our business and results of operations, including the imposition of substantial fines or other sanctions.
We may expand our business through the acquisition of companies or businesses or by entering into collaborations or by in-licensing product candidates, each of which could disrupt our business and harm our financial condition.
We have in the past and may in the future seek to expand our pipeline and capabilities by acquiring one or more companies or businesses, entering into collaborations, or in-licensing one or more product candidates. Acquisitions, collaborations and in-licenses involve numerous risks, including, but not limited to:
|
● |
substantial cash expenditures; |
|
● |
technology development risks; |
|
● |
potentially dilutive issuances of equity securities; |
|
● |
incurrence of debt and contingent liabilities, some of which may be difficult or impossible to identify at the time of acquisition; |
|
● |
difficulties in assimilating the operations of the acquired companies; |
|
● |
potential disputes regarding contingent consideration; |
|
● |
diverting our management’s attention away from other business concerns; |
|
● |
entering markets in which we have limited or no direct experience; and |
|
● |
potential loss of our key employees or key employees of the acquired companies or businesses. |
We cannot provide assurance that any acquisition, collaboration, or in-license will result in short-term or long-term benefits to us. We may incorrectly judge the value or worth of an acquired company or business or in-licensed product candidate. In addition, our future success would depend in part on our ability to manage the growth associated with some of these acquisitions, collaborations and in-licenses. We cannot provide assurance that we would be able to successfully combine our business with that of acquired businesses, manage a collaboration or integrate in-licensed product candidates. Furthermore, the development or expansion of our business may require a substantial capital investment by us.
Our current or future product candidates may cause undesirable side effects or have other properties that could prevent their regulatory approval, limit the commercial scope of their approved uses, or result in significant negative consequences following any marketing approval.
Undesirable side effects caused by our product candidates could cause us or regulatory authorities to delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign regulatory authorities. Results of our trials could reveal unacceptable side effects or unexpected characteristics. In such an event, we could suspend or terminate our clinical trials or the FDA or comparable foreign regulatory authorities could order us to cease clinical trials or deny approval of our product candidates for any or all targeted indications. Drug-related side effects could affect patient recruitment or the ability of enrolled subjects to complete the trial or result in potential product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly.
Additionally, if one or more of our product candidates receives marketing approval, and we or others later identify undesirable side effects caused by any such products, a number of potentially significant negative consequences could result, including:
|
● |
we may suspend marketing of, or withdraw or recall, such product; |
|
● |
regulatory authorities may withdraw approvals of such product; |
|
● |
regulatory authorities may require additional warnings on the label or otherwise seek to limit the scope of the approved uses reflected in the label of such product; |
|
● |
the FDA may require the use of or modification of a Risk Evaluation and Mitigation Strategy (“REMS”) or a comparable foreign regulatory authority may require the establishment or modification of a similar strategy that may, for instance, restrict distribution of our products and impose other implementation requirements on us; |
|
● |
regulatory authorities may require that we conduct post-marketing studies; |
|
● |
we could be sued and held liable for harm caused to patients; and |
|
● |
our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the particular product candidate or otherwise materially harm the commercial prospects for the product candidate, if approved, and could significantly harm our business, results of operations and prospects.
We face the risk of product liability claims, which could exceed our insurance coverage and produce recalls, each of which could deplete our cash resources.
We are exposed to the risk of product liability claims alleging that use of our product candidates caused an injury or harm. These claims can arise at any point in the development, testing, manufacture, marketing, or sale of our product candidates and may be made directly by patients involved in clinical trials of our product candidates, by consumers or healthcare providers, or by individuals, organizations, or companies selling our products. Product liability claims can be expensive to defend, even if the product or product candidate did not actually cause the alleged injury or harm.
Insurance covering product liability claims becomes increasingly expensive as a product candidate moves through the development pipeline to commercialization. To protect against potential product liability risks, we have AUD$20 million per occurrence and AUD$20 million aggregate clinical trial insurance for the REMEDY Phase II clinical trial in Australia and US$5.0 million product liability insurance coverage. However, there can be no assurance that such insurance coverage is or will continue to be adequate or available to us at a cost acceptable to us or at all. We may choose or find it necessary under our collaboration agreements to increase our insurance coverage in the future. We may not be able to secure greater or broader product liability insurance coverage on acceptable terms or at reasonable costs when needed. Any liability for damages resulting from a product liability claim could exceed the amount of our coverage, require us to pay a substantial monetary award from our own cash resources and have a material adverse effect on our business, financial condition, and results of operations. Moreover, a product recall, if required, could generate substantial negative publicity about our products and business, inhibit or prevent commercialization of other products and product candidates, or negatively impact existing or future collaborations.
If we are unable to maintain product liability insurance required by our third parties, the corresponding agreements would be subject to termination, which could have a material adverse impact on our operations.
Some of our license, clinical trials and other agreements with third parties require, and in the future may require, us to maintain product liability insurance. If we cannot maintain acceptable amounts of coverage on commercially reasonable terms in accordance with the terms set forth in these agreements, the corresponding agreements would be subject to termination, which could have a material adverse impact on our operations.
A risk of product liability claims, and related negative publicity, is inherent in the development of human therapeutics and other products. Product liability insurance is expensive, its availability is limited, and it may not be offered on terms acceptable to us, or at all. The commercialization of our potential products could be inhibited or prevented by an inability to maintain sufficient insurance coverage on reasonable terms or to otherwise protect against potential product liability claims. A product liability claim against us or the withdrawal of a product from the market could have a material adverse effect upon us and our financial condition.
A variety of risks are associated with operating our business internationally which could materially adversely affect our business.
We conduct certain R&D operations in Australia. In addition, we may conduct certain future clinical trials and plan to seek regulatory approval of our product candidates outside of the United States. Accordingly, we are subject to risks related to operating in foreign countries, including:
|
● |
different regulatory requirements for drug approvals in foreign countries; |
|
● |
different standards of care in various countries that could complicate the evaluation of our product candidates; |
|
● |
different United States and foreign drug import and export rules; |
|
● |
reduced protection for intellectual property rights in certain countries; |
|
● |
unexpected changes in tariffs, trade barriers, and regulatory requirements; |
|
● |
different reimbursement systems and different competitive drugs indicated to treat the indications for which our product candidates are being developed; |
|
● |
economic weakness, including inflation, or political instability in particular foreign economies and markets; |
|
● |
compliance with tax, employment, immigration, and labor laws for employees living or traveling abroad; |
|
● |
compliance with the Foreign Corrupt Practices Act and other anti-corruption and anti-bribery laws; |
|
● |
foreign taxes, including withholding of payroll taxes; |
|
● |
foreign currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country; |
|
● |
workforce uncertainty in countries where labor unrest is more common than in the United States; |
|
● |
production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; |
|
● |
potential liability resulting from development work conducted by foreign partners; |
|
● |
business interruptions resulting from natural disasters or geopolitical actions, including war and terrorism, or systems failure including cybersecurity breaches; and |
|
● |
compliance with evolving and expansive international data privacy laws, such as the European Union General Data Protection Regulation. |
Future legislation in the United States, Europe or other countries, and/or regulations and policies adopted by the FDA, the EMA or comparable regulatory authorities, may increase the time and cost required for us or our collaborator to conduct and complete clinical trials of our current or future product candidates.
The FDA and the EMA have each established regulations to govern the product development and approval process, as have other foreign regulatory authorities. The policies of the FDA, the EMA and other regulatory authorities may change. For example, in December 2016, the 21st Century Cures Act (“Cures Act”) was signed into law. The Cures Act, among other things, is intended to modernize the regulation of drugs and spur innovation, but not all of its provisions have yet been implemented. Additionally, in August 2017, the FDA issued final guidance setting forth its current thinking with respect to development programs and clinical trial designs for antibacterial drugs to treat serious bacterial diseases in patients with an unmet medical need. We cannot predict what if any effect the Cures Act or any existing or future guidance from the FDA or other regulatory authorities will have on the development of our product candidates.
Recently enacted and future legislation may increase the difficulty and cost for us and our collaborators to obtain marketing approval of and commercialize our product candidates and affect the prices we may obtain.
In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any product candidates for which we obtain marketing approval.
Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. For example, the ACA, which was enacted in the United States in March 2010, includes measures to change health care delivery, decrease the number of individuals without insurance, ensure access to certain basic health care services, and contain the rising cost of care. This healthcare reform movement, including the enactment of the ACA, has significantly changed health care financing by both governmental and private insurers in the United States. With respect to pharmaceutical manufacturers, the ACA increased the number of individuals with access to health care coverage, including prescription drug coverage, but it simultaneously imposed, among other things, increased liability for rebates and discounts owed to certain entities and government health care programs, new fees for the manufacture or importation of certain branded drugs, and new transparency reporting requirements under the Physician Payments Sunshine Act.
Since its enactment, there have been judicial and Congressional challenges to certain aspects of the ACA, as well as recent efforts by the current administration to repeal or replace certain aspects of the ACA. Since January 2017, two U.S. Presidential Executive Orders have been signed and other directives designed to delay the implementation of any certain provisions of the ACA or otherwise remove some of the requirements for health insurance mandated by the ACA. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the ACA. While Congress has not passed comprehensive repeal legislation, two bills affecting the implementation of certain taxes under the ACA have been signed into law. The Tax Cuts and Jobs Act of 2017 includes a provision repealing, effective January 1, 2019, the tax imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the "individual mandate." Additionally, on January 22, 2018, the U.S. President signed a continuing resolution on appropriations for fiscal year 2018 that delayed the implementation of certain ACA-mandated fees, including the tax on certain high cost employer-sponsored insurance plans, the annual fee imposed on certain health insurance providers based on market share, and the medical device excise tax on non-exempt medical devices. Further, the Bipartisan Budget Act of 2018, or the “BBA,” among other things, amends the ACA, effective January 1, 2019, to increase from 50 percent to 70 percent the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D and to close the coverage gap in most Medicare drug plans, commonly referred to as the "donut hole."
In addition to the ACA, other federal health reform measures have been proposed and adopted in the United States. For example, legislation has been enacted to reduce the level of reimbursement paid to providers under the Medicare program over time, as well as phase in alternative payment models for provider services under the Medicare program with the goal of incentivizing the attainment of pre-defined quality measures. As these measures are not fully in effect, and since the U.S. Congress could intervene to prevent their full implementation, it is unclear how payment reductions or the introduction of the quality payment program will impact overall physician reimbursement under the Medicare program. It is also unclear if changes in Medicare payments to providers would impact such providers' willingness to prescribe and administer our products, if approved. Further, there has been heightened governmental scrutiny over the manner in which companies set prices for their marketed products. For example, there have been several recent Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and patient programs, and reform government program reimbursement methodologies for drug products.
We expect that the ACA, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we may receive for any product, if approved. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our drugs.
Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for drugs. We cannot be sure whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our current or future product candidates, if any, may be. In addition, increased scrutiny by the U.S. Congress of the FDA's approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
We conduct certain research and development operations through our Australian wholly-owned subsidiary. If we lose our ability to operate in Australia, or if our subsidiary is unable to receive the research and development incentive payment allowed by Australian regulations, our business and results of operations could suffer.
In July 2016, we formed a wholly-owned Australian subsidiary, DiaMedica Australia Pty Ltd, to conduct various clinical activities for our product and development candidate in Australia. Due to the geographical distance and lack of employees currently in Australia, as well as our lack of experience operating in Australia, we may not be able to efficiently or successfully monitor, develop and commercialize our lead product candidate in Australia, including conducting clinical trials. Furthermore, we have no assurance that the results of any clinical trials that we conduct for our product candidate in Australia will be accepted by the FDA or foreign regulatory authorities for development and commercialization approvals.
In addition, current Australian tax regulations provide for a refundable R&D incentive payment equal to 43.5% of qualified expenditures. We received incentive payments of approximately AUD$ 306,000 and AUD$ 777,000 during 2017 and 2018, respectively, for research expenditures made during 2016 and 2017. If our subsidiary loses its ability to operate in Australia, or if we are ineligible or unable to receive the R&D incentive payment, or the Australian government significantly reduces or eliminates the incentive program, our business and results of operation may be adversely affected.
Risks Related to Intellectual Property
If we are unable to adequately protect and enforce our intellectual property, our competitors may take advantage of our development efforts or acquired technology and compromise our prospects of marketing and selling our key product candidates.
We believe that patents and other proprietary rights are key to our business. Our policy is to file patent applications to protect technology, inventions, and improvements that may be important to the development of our business. We also rely upon trade secrets, know-how, continuing technological innovations, and licensing opportunities to develop and maintain our competitive position. We plan to enforce our issued patents and our rights to proprietary information and technology. We review third-party patents and patent applications, both to refine our own patent strategy and to identify useful licensing opportunities.
Our success depends, in part, on our ability to secure and protect our intellectual property rights and to operate without infringing on the proprietary rights of others or having third parties circumvent the rights owned or licensed by us. We have a number of patents, patent applications and rights to patents related to our compounds, product candidates and technology, but we cannot be certain that they will be enforceable or provide adequate protection or that pending patent applications will result in issued patents.
To the extent that development, manufacturing, and testing of our product candidates is performed by third party contractors, such work is performed pursuant to fee for service contracts. Under the contracts, all intellectual property, technology know-how, and trade secrets arising under such agreements are our exclusive property and must be kept confidential by the contractors. It is not possible for us to be certain that we have obtained from the contractors all necessary rights to such technologies. Disputes may arise as to the scope of the contract or possible breach of contract. No assurance can be given that our contracts would be enforceable or would be upheld by a court.
The patent positions of pharmaceutical and biotechnology firms, ourselves included, are uncertain and involve complex questions of law and fact for which important legal issues remain unresolved. Therefore, it is not clear whether our pending patent applications will result in the issuance of patents or whether we will develop additional proprietary products which are patentable. Part of our strategy is based on our ability to secure a patent position to protect our technology. There is no assurance that we will be successful in this approach and failure to secure patent protection may have a material adverse effect upon us and our financial condition. Also, we may fail in our attempt to commercialize products using currently patented or licensed technology without having to license additional patents. Moreover, it is not clear whether the patents issued or to be issued will provide us with any competitive advantages or if any such patents will be the target of challenges by third parties, whether the patents of others will interfere with our ability to market our products, or whether third parties will circumvent our patents by means of alternate processes. Furthermore, it is possible for others to develop products which have the same effect as our product candidates or technologies on an independent basis or to design around technologies patented by us. Patent applications relating to or affecting our business may have been filed by pharmaceutical or biotechnology companies or academic institutions. Such applications may conflict with our technologies or patent applications and such conflict could reduce the scope of patent protection which we could otherwise obtain or even lead to the rejection of our patent applications. There is no assurance that we can enter into licensing arrangements on commercially reasonable terms, or develop or obtain alternative technology in respect of, patents issued to third parties that incidentally cover our products or production technologies. Any inability to secure licenses or alternative technology could result in delays in the introduction of some of our product candidates or even lead to us being prevented from pursuing the development, manufacture, or sale of certain products. Moreover, we could potentially incur substantial legal costs in defending legal actions which allege patent infringement, or by initiating patent infringement suits against others. It is not possible for us to be certain that we are the creator of inventions covered by pending patent applications or that we were the first to invent or file patent applications for any such inventions. While we have used commercially reasonable efforts to obtain assignments of intellectual property from all individuals who may have created materials on our behalf (including with respect to inventions covered by our patent and pending patent applications), it is not possible for us to be certain that we have obtained all necessary rights to such materials. No assurance can be given that our patents, if issued, would be upheld by a court, or that a competitor’s technology or product would be found to infringe on our patents. Moreover, much of our technology know-how that is not patentable may constitute trade secrets. Therefore, we require our employees, consultants, advisors, and collaborators to enter into confidentiality agreements either as stand-alone agreements or as part of their consulting contracts. However, no assurance can be given that such agreements will provide meaningful protection of our trade secrets, know-how, or other proprietary information in the event of any unauthorized use or disclosure of confidential information. Also, while we have used commercially reasonable efforts to obtain executed copies of such agreements from all employees, consultants, advisors and collaborators, no assurance can be given that executed copies of all such agreements have been obtained.
We may require additional third-party licenses to effectively develop and manufacture our key products and are currently unable to predict the availability or cost of such licenses.
A substantial number of patents have already been issued to other biotechnology and pharmaceutical companies. To the extent that valid third-party patent rights cover our product candidates, we or our strategic collaborators would be required to seek licenses from the holders of these patents in order to manufacture, use, or sell these product candidates, and payments under them would reduce our profits from these product candidates. We are currently unable to predict the extent to which we may wish or be required to acquire rights under such patents, the availability and cost of acquiring such rights, and whether a license to such patents will be available on acceptable terms, or at all. There may be patents in the U.S. or in foreign countries or patents issued in the future that are unavailable to license on acceptable terms. Our inability to obtain such licenses may hinder or eliminate our ability to develop, manufacture and market our product candidates.
Changes in patent law and its interpretation could diminish the value of patents in general, thereby impairing our ability to protect our product candidates.
As is the case with other biotechnology and pharmaceutical companies, our success is heavily dependent on intellectual property rights, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves technological and legal complexity, and obtaining and enforcing biopharmaceutical patents is costly, time consuming, and inherently uncertain. The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our and our licensors’ or collaborators’ ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts, and the U.S. Patent and Trademark Office (“USPTO”), the laws and regulations governing patents could change in unpredictable ways that would weaken our and our licensors’ or collaborators’ ability to obtain new patents or to enforce existing patents and patents we and our licensors or collaborators may obtain in the future. Changes in either the patent laws or interpretation of the patent laws in the United States or other countries could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents.
Assuming that other requirements for patentability are met, prior to March 2013, in the United States, the first to invent the claimed invention was entitled to the patent, while outside the United States, the first to file a patent application was entitled to the patent. After March 2013, under the Leahy-Smith America Invents Act, or the America Invents Act, enacted in September 2011, the United States transitioned to a first inventor to file system in which, assuming that other requirements for patentability are met, the first inventor to file a patent application will be entitled to the patent on an invention regardless of whether a third party was the first to invent the claimed invention. A third party that files a patent application in the USPTO after March 2013, but before us could therefore be awarded a patent covering an invention of ours even if we had made the invention before it was made by such third party. This will require us to be cognizant of the time from invention to filing of a patent application. Since patent applications in the United States and most other countries are confidential for a period of time after filing or until issuance, we cannot be certain that we or our licensors were the first to either (i) file any patent application related to our product candidates or (ii) invent any of the inventions claimed in our or our licensor’s patents or patent applications.
The America Invents Act also includes a number of significant changes that affect the way patent applications will be prosecuted and also may affect patent litigation. These include allowing third party submission of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent by USPTO administered post-grant proceedings, including post-grant review, inter partes review, and derivation proceedings. Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in United States federal courts necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third party may attempt to use the USPTO procedures to invalidate our patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action. Therefore, the America Invents Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our owned or in-licensed patent applications and the enforcement or defense of our owned or in-licensed issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations, and prospects.
The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our and our licensors’ or collaborators’ ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts, and the U.S. Patent and Trademark Office, and similar legislative, judicial, and administrative bodies in other countries, the laws and regulations governing patents could change in unpredictable ways that would weaken our and our licensors’ or collaborators’ ability to obtain new patents or to enforce existing patents and patents we and our licensors or collaborators may obtain in the future.
Litigation regarding patents, patent applications, and other proprietary rights may be expensive, time consuming and cause delays in the development and manufacturing of our key product candidates.
Third parties may claim that we are using their proprietary information without authorization. Third parties may also have or obtain patents and may claim that technologies licensed to or used by us infringe their patents. If we are required to defend patent infringement actions brought by third parties, or if we sue to protect our own patent rights or otherwise to protect our proprietary information and to prevent its disclosure, we may be required to pay substantial litigation costs and managerial attention may be diverted from business operations even if the outcome is in our favor. In addition, any legal action that seeks damages or an injunction to stop us from carrying on our commercial activities relating to the affected technologies could subject us to monetary liability (including treble damages and attorneys’ fees if we are found to have willfully infringed) and require us or any third-party licensors to obtain a license to continue to use the affected technologies. We cannot predict whether we would prevail in any of these types of actions or that any required license would be available on commercially acceptable terms or at all. Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources.
Competitors may infringe our patents or other intellectual property. If we were to initiate legal proceedings against a third party to enforce a patent covering our product candidates, the defendant could counterclaim that the patent covering our product candidate is invalid and/or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness, written description or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. The outcome following legal assertions of invalidity and unenforceability is unpredictable. Moreover, similar challenges may be made by third parties outside the context of litigation, e.g., via administrative proceedings such as post grant or inter partes review in the United States or via oppositions or other similar proceedings in other countries/regions.
Interference or derivation proceedings provoked by third parties or brought by us or declared by the USPTO may be necessary to determine the priority of inventions with respect to our patents or patent applications. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms or at all, or if a non-exclusive license is offered and our competitors gain access to the same technology. Our defense of litigation, validity or enforceability, interference or derivation proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. In addition, the uncertainties associated with litigation or such other proceedings could have a material adverse effect on our ability to raise the funds necessary to continue our clinical trials, continue our research programs, license necessary technology from third parties, or enter into development partnerships that would help us bring our product candidates to market.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation or administrative proceedings, there is a risk that some of our confidential information could be compromised by disclosure. There could also be public announcements of the results of hearings, motions, or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our common shares.
If we fail to comply with our obligations in the agreements under which we license intellectual property rights from third parties or otherwise experience disruptions to our business relationships with our licensors, we could lose intellectual property rights that are important to our business.
We are a party to a license agreement relating to an expression system and cell line for use in the production of DM199 or any human KLK1, and we may need to obtain additional licenses from others to advance our R&D activities or allow the commercialization of DM199 or any other product candidates we may identify and pursue. Future license agreements may impose, various development, diligence, commercialization, and other obligations on us. If any of our in-licenses are terminated, or if the underlying patents fail to provide the intended exclusivity, competitors or other third parties main gain access to technologies that are material to our business, and we may be required to cease our development and commercialization of DM199 or other product candidates that we may identify or to seek alternative manufacturing methods. However, suitable alternatives may not be available or the development of suitable alternatives may result in a significant delay in our commercialization of DM199. Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations, and prospects.
Moreover, disputes may arise regarding intellectual property subject to a licensing agreement, including, but not limited to:
|
● |
the scope of rights granted under the license agreement and other interpretation-related issues; |
|
● |
the extent to which our product candidates, technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement; |
|
● |
the sublicensing of patent and other rights under our collaborative development relationships; |
|
● |
our diligence obligations under the license agreement and what activities satisfy those diligence obligations; |
|
● |
the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners; and |
|
● |
the priority of invention of patented technology. |
In addition, the agreements under which we currently license intellectual property or technology from third parties are complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations, and prospects. Moreover, if disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on commercially acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates, which could have a material adverse effect on our business, financial conditions, results of operations, and prospects.
Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them.
Because we rely on third parties to develop our products, we must share trade secrets with them. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, consulting agreements, or other similar agreements with our collaborators, advisors, employees, and consultants prior to beginning research or disclosing proprietary information. These agreements typically restrict the ability of our collaborators, advisors, employees, and consultants to publish data potentially relating to our trade secrets. Our academic collaborators typically have rights to publish data, provided that we are notified in advance and may delay publication for a specified time in order to secure our intellectual property rights arising from the collaboration. In other cases, publication rights are controlled exclusively by us, although in some cases we may share these rights with other parties. We also conduct joint R&D programs which may require us to share trade secrets under the terms of R&D collaboration or similar agreements. However, we cannot be certain that such agreements have been entered into with all relevant parties. Moreover, despite our efforts to protect our trade secrets, our competitors may discover our trade secrets, either through breach of these agreements, independent development, or publication of information including our trade secrets in cases where we do not have proprietary or otherwise protected rights at the time of publication. Trade secrets can be difficult to protect. If the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient recourse against third parties for misappropriating any trade secrets. A competitor’s discovery of our trade secrets may impair our competitive position and could have a material adverse effect on our business and financial condition.
Patent terms may be inadequate to protect our competitive position on our product candidates for an adequate amount of time.
Patents have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date. Various extensions may be available, but the life of a patent, and the protection it affords, is limited. Even if patents covering our product candidates are obtained, once the patent life has expired, we may be open to competition from competitive products, including generics or biosimilars. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties or that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology and pharmaceutical industry, we employ individuals who were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees, consultants and independent contractors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of any of our employee’s former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel, which could adversely impact our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or applications will be due to be paid to the USPTO and various governmental patent agencies outside of the United States in several stages over the lifetime of the patents and/or applications. We have systems in place to remind us to pay these fees, and we employ an outside firm and rely on our outside counsel to pay these fees due to non-U.S. patent agencies. The USPTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. We employ reputable law firms and other professionals to help us comply, and in many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, there are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, our competitors might be able to enter the market and this circumstance would have a material adverse effect on our business.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on our product candidates in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States can be less extensive than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and may also export infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our products and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets, and other intellectual property protection, particularly those relating to biotechnology products, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions, whether or not successful, could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
Risks Related to Our Common Shares and this Offering
Our management will have broad discretion and flexibility as to how to use the net proceeds from this offering and may use the net proceeds in ways with which you disagree or which may not prove effective.
We currently intend to use the net proceeds from this offering as discussed under “Use of Proceeds” in this prospectus. We have not allocated specific amounts of the net proceeds from this offering for any of the purposes set forth in that section. Accordingly, our management will have significant discretion and flexibility in applying the net proceeds of this offering. You will be relying on the judgment of our management with regard to the use of these net proceeds, and you will not have the opportunity, as part of your investment decision, to assess whether the net proceeds are being used appropriately. It is possible that the net proceeds will be invested in a way that does not yield a favorable, or any, return for us. The failure of our management to use such funds effectively could have a material adverse effect on our business, financial condition, operating results and cash flow.
Purchasers of common shares in this offering will experience immediate and substantial dilution in the book value of their investment. You may experience further dilution upon exercise of our outstanding options and warrants.
The initial public offering price per common share in this offering is substantially higher than the net tangible book value per common share before giving effect to this offering. Accordingly, if you purchase common shares in this offering, you will incur immediate substantial dilution of approximately $ per share, representing the difference between the assumed initial public offering price of $ per common share, which is the midpoint of the price range set forth on the cover page of this prospectus, and our as adjusted net tangible book value per share as of September 30, 2018. In addition, if outstanding options or warrants are exercised, you could experience further dilution. For a further description of the dilution that you will experience immediately after this offering, see the section in this prospectus entitled “Dilution.”
Our recent share consolidation may not have the intended benefits.
On November 15, 2018, we implemented a share consolidation of our common shares, which was previously approved by our shareholders, pursuant to which each 20 common shares outstanding on the record date for the share consolidation was combined into one common share. We cannot predict whether the share consolidation will increase the market price for our common shares on a sustained basis. The history of similar share consolidations for companies in similar circumstances is varied, and we cannot predict whether:
|
● |
the share consolidation will result in a sustained price per share that will attract brokers and investors who do not trade in lower priced stocks; |
|
● |
the share consolidation will result in a price per share that will increase our ability to attract and retain employees and other service providers; |
|
● |
the market price per share will remain at a level in excess of the minimum bid price as required for continued listing on The Nasdaq Capital Market; or |
|
● |
even if the share consolidation does increase the market price of our common shares on a sustained basis, we will otherwise meet the requirements of The Nasdaq Capital Market and be able to maintain our listing. |
Our common share price has been volatile in recent years and may continue to be volatile.
Our common shares trade in Canada on the TSX Venture Exchange under the trading symbol “DMA” and over-the-counter in the United States on the OTCQB marketplace under the trading symbol “DMCAF.” We have applied to list our common shares on The Nasdaq Capital Market under the trading symbol “DMAC.” A number of factors could influence the volatility in the trading price of our common shares, including changes in the economy or in the financial markets, industry related developments, and the impact of material events and changes in our operations. Our quarterly losses may vary because of expenses we incur related to future research including the timing of costs for manufacturing and initiating and completing preclinical and clinical trials. Each of these factors could lead to increased volatility in the market price of our common shares. In addition, the market prices of the securities of our competitors may also lead to fluctuations in the trading price of our common shares. As a result of this volatility, you may not be able to sell your common shares at or above the initial public offering price.
We do not have a very active trading market for our common shares and one may never develop.
Our common shares trade in Canada on the TSX Venture Exchange under the trading symbol “DMA” and over-the-counter in the United States on the OTCQB marketplace under the trading symbol “DMCAF.” We have applied to list our common shares on The Nasdaq Capital Market under the trading symbol “DMAC.” We do not have a very active trading market for our common shares and one may never develop, even after this offering. Although we anticipate that our common shares will be approved for listing on The Nasdaq Capital Market and a more active trading market for our shares will develop after this offering, we can give no assurance that this will occur or that an active trading market will be sustained following this offering. If an active market for our common shares does not develop, it may be difficult for you to sell shares you purchase in this offering at a favorable price or at all.
We have never paid dividends and do not expect to do so in the foreseeable future.
We have not declared or paid any cash dividends on our common shares to date. The payment of dividends in the future will be dependent on our earnings and financial condition and on such other factors as our Board of Directors considers appropriate. Unless and until we pay dividends, shareholders may not receive a return on their shares. There is no present intention by our Board of Directors to pay dividends on our common shares. We currently intend to retain all of our future earnings, if any, to finance the growth and development of our business. In addition, the terms of any future debt agreements may preclude us from paying dividends. As a result, appreciation, if any, in the market price of our common shares will be your sole source of gain for the foreseeable future.
We may issue additional common shares resulting in share ownership dilution.
Future dilution may occur due to additional future equity financing events by us. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interests of our shareholders will be diluted. In addition, if outstanding options, warrants, or deferred share units are exercised or otherwise converted into our common shares, our shareholders will experience additional dilution.
It may be difficult for non-Canadian shareholders or other investors to obtain and enforce judgments against us because of our Canadian incorporation and presence.
We are a corporation existing under the federal laws of Canada. Two of our directors and several of the experts we utilize are residents of Canada, and all or a substantial portion of their assets, and a portion of our assets, are located outside the United States. Consequently, it may be difficult for holders of our securities who reside in the United States to effect service within the United States upon those directors and the experts who are not residents of the United States. It may also be difficult for holders of our securities who reside in the United States to realize in the United States upon judgments of courts of the United States predicated upon our civil liability and the civil liability of our directors, officers, and experts under the United States federal securities laws. Our shareholders and other investors should not assume that Canadian courts (i) would enforce judgments of United States courts obtained in actions against us or such directors, officers, or experts predicated upon the civil liability provisions of the United States federal securities laws or the securities or “blue sky” laws of any state or jurisdiction of the United States, or (ii) would enforce, in original actions, liabilities against us or such directors, officers, or experts predicated upon the United States federal securities laws or any securities or “blue sky” laws of any state or jurisdiction of the United States. In addition, the protections afforded by Canadian securities laws may not be available to our shareholders or other investors in the United States.
If there are substantial sales of our common shares, the market price of our common shares could decline.
Sales of substantial numbers of our common shares could cause a decline in the market price of our common shares. Any sales by existing shareholders or holders who exercise their warrants or stock options may have an adverse effect on our ability to raise capital and may adversely affect the market price of our common shares.
Our common shares trade on more than one market and this may result in price variations.
Our common shares trade in Canada on the TSX Venture Exchange under the trading symbol “DMA” and over-the-counter in the United States on the OTCQB marketplace under the trading symbol “DMCAF.” We have applied to list our common shares on The Nasdaq Capital Market under the trading symbol “DMAC.” Trading in our common shares on these markets takes place in different currencies (U.S. dollars for OTCQB marketplace and The Nasdaq Capital Market and Canadian dollars on the TSX Venture Exchange) and at different times (due to different time zones, trading days and public holidays in the U.S. and Canada). The trading prices of our common shares on these two markets may differ due to these and other factors. Any decrease in the trading price of our common shares on one of these markets could cause a decrease in the trading price of our common shares on the other market. Differences in trading prices on the two markets could negatively impact our trading price.
We could be subject to securities class action litigation, which is expensive and could divert management attention.
In the past, securities class action litigation has often been brought against a company following a decline or increase in the market price of its securities or certain significant business transactions. We may become involved in this type of litigation in the future. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and our resources, which could harm our business.
If securities or industry analysts do not publish research or reports about our business, or publish negative reports about our business, the market stock of our common shares and trading volume could decline.
The trading market for our common shares in the United States after this offering will depend in part on the research and reports that securities or industry analysts publish about us or our business. We do not have any control over these analysts. There can be no assurance that analysts will cover us or provide favorable coverage. If one or more of the analysts who cover us downgrade our shares or change their opinion of our shares, our share price would likely decline. If one or more of these analysts cease coverage of our company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which could cause the market price of our common shares or trading volume to decline.
We are an “emerging growth company,” and the reduced disclosure requirements applicable to emerging growth companies may make our common shares less attractive to our shareholders and other investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012. We may remain an emerging growth company until the last day of the fiscal year following the fifth anniversary of our first sale of common shares pursuant to a registration statement under the Securities Act of 1933, as amended, or the “Securities Act,” until such earlier time as we have more than $1.07 billion in annual revenue, the market value of our common shares held by non-affiliates is more than $700 million or we issue more than $1 billion of non-convertible debt over a three-year period. For so long as we remain an emerging growth company, we are permitted and intend to rely on exemptions from certain disclosure requirements that are applicable to other public companies that are not emerging growth companies. These exemptions include not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002 (“Section 404”) not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements, reduced disclosure obligations regarding executive compensation and exemptions from the requirements of holding a non-binding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. Our shareholders and other investors may find our common shares less attractive as a result of our reliance on these exemptions. If some of our shareholders or other investors find our common shares less attractive as a result, there may be a less active trading market for our common shares and the trading price of our common shares may be more volatile.
In addition, Section 107 of the JOBS Act provides that an emerging growth company can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised financial accounting standards. An emerging growth company can therefore delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. However, we have determined to opt out of such extended transition period and, as a result, we will comply with new or revised financial accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that our decision to opt out of the extended transition period for complying with new or revised financial accounting standards is irrevocable.
Our shareholders and other investors may find our common shares less attractive as a result of our reliance on these exemptions. If some of our shareholders or other investors find our common shares less attractive as a result, there may be a less active trading market for our common shares and the trading price of our common shares may be more volatile.
We will incur increased costs as a result of operating as a U.S. public reporting company and maintaining a dual listing on The Nasdaq Capital Market and the TSX Venture Exchange, and our management is required to devote substantial time to new compliance initiatives.
As a U.S. public reporting company, we anticipate that we will incur, particularly after we are no longer an “emerging growth company,” significant legal, accounting and other expenses that we did not incur as a company listed solely on the TSX Venture Exchange. In addition, the Sarbanes-Oxley Act of 2002 and rules subsequently implemented by the SEC and Nasdaq have imposed various requirements on U.S. public companies, including establishment and maintenance of effective disclosure and financial controls and corporate governance practices. We may have to hire additional accounting, finance, and other personnel to assist us with becoming a U.S. public reporting company, and our efforts to comply with U.S. public company reporting requirements, and our management and other personnel will need to devote a substantial amount of time towards maintaining compliance with these requirements. These requirements increase our legal and financial compliance costs and will make some activities more time-consuming and costly.
We have no operating experience as a publicly traded company in the U.S.
We have no operating experience as a publicly traded company in the U.S. Although the individuals who now constitute our management team have experience managing a publicly-traded company, there is no assurance that the past experience of our management team will be sufficient to operate the Company as a publicly traded company in the United States, including timely compliance with the disclosure requirements of the SEC. Following the completion of this offering, we will be required to develop and implement internal control systems and procedures in order to satisfy the periodic and current reporting requirements under applicable SEC regulations and comply with the Nasdaq listing standards. These requirements will place significant strain on our management team, infrastructure and other resources. In addition, our management team may not be able to successfully or efficiently manage the Company as a U.S. public reporting company that is subject to significant regulatory oversight and reporting obligations.
Our inability to comply with Nasdaq’s continued listing requirements could result in our common shares being delisted, which could affect the market price and liquidity of our common shares and reduce our ability to raise capital.
Upon completion of this offering, we will be required to meet certain qualitative and financial tests to maintain the listing of our common shares on The Nasdaq Capital Market. If we do not maintain compliance with Nasdaq’s continued listing requirements within specified periods and subject to permitted extensions, our common shares may be recommended for delisting (subject to any appeal we would file). No assurance can be provided that we will comply with these continued listing requirements. If our common shares were delisted, it could be more difficult to buy or sell our common shares and to obtain accurate quotations, and the price of our common shares could suffer a material decline. Delisting would also impair our ability to raise capital.
Our shareholder rights plan may delay or prevent an acquisition of us that shareholders may consider favorable or may prevent efforts by our shareholders to change our directors or our management, which could decrease the value of your common shares.
Our shareholders approved the adoption of a shareholder rights plan agreement on December 21, 2017. The shareholder rights plan is designed to provide adequate time for our Board of Directors and shareholders to assess an unsolicited takeover bid for our company, to provide our Board of Directors with sufficient time to explore and develop alternatives for maximizing shareholder value if a takeover bid is made, and to provide shareholders with an equal opportunity to participate in a takeover bid and receive full and fair value for their common shares. The shareholder rights plan is set to expire at the close of our annual meeting of shareholders in 2020. The rights will become exercisable only when a person, including any party related to it, acquires or attempts to acquire 20% or more of our outstanding common shares without complying with the “permitted bid” provisions of the plan or without approval of our Board of Directors. Should such an acquisition occur or be announced, each right would, upon exercise, entitle a rights holder, other than the acquiring person and related persons, to purchase common shares at a 50% discount to the market price at the time. Under the plan, a “permitted bid” is a bid made to all holders of our common shares and which is open for acceptance for not less than 60 days. If at the end of 60 days at least 50% of the outstanding common shares, other than those owned by the offeror and certain related parties have been tendered, the offeror may take up and pay for the common shares but must extend the bid for a further 10 days to allow other shareholders to tender.
While we believe our rights plan enables our Board of Directors to help ensure that our shareholders are not deprived of the opportunity to realize the full and fair value of their investments, the rights plan may inhibit a change in control of our company by a third party in a transaction not approved by our Board of Directors. If a change in control is inhibited or delayed in this manner, it may adversely affect the market price of our common shares.
Any failure to maintain an effective system of internal controls may result in material misstatements of our consolidated financial statements or cause us to fail to meet our reporting obligations or fail to prevent fraud; and in that case, our shareholders or other investors could lose confidence in our financial reporting, which would harm our business and could negatively impact the price of our common shares.
Effective internal controls are necessary for us to provide reliable financial reports and prevent fraud. If we fail to maintain an effective system of internal controls, we might not be able to report our financial results accurately or prevent fraud; and in that case, our shareholders or other investors could lose confidence in our financial reporting, which would harm our business and could negatively impact the price of our common shares. As a result of our limited administrative staffing levels, internal controls which rely on segregation of duties in many cases are not possible. Due to resource constraints and the present stage of our development, we do not have sufficient size and scale to warrant the hiring of additional staff to address this potential weakness at this time. To help mitigate the impact of this, we are highly reliant on the performance of compensating procedures and senior management’s review and approval. Even if we conclude that our internal control over financial reporting provides reasonable assurance regarding the reliability of financial reporting and the preparation of consolidated financial statements for external purposes in accordance with GAAP, because of its inherent limitations, internal control over financial reporting may not prevent or detect fraud or misstatements. Failure to implement required new or improved controls, or difficulties encountered in their implementation, could harm our results of operations or cause us to fail to meet our future reporting obligations.
If we fail to timely achieve and maintain the adequacy of our internal control over financial reporting, we may not be able to produce reliable financial reports or help prevent fraud. Our failure to achieve and maintain effective internal control over financial reporting could prevent us from complying with our reporting obligations on a timely basis, which could result in the loss of shareholder or other investor confidence in the reliability of our consolidated financial statements, harm our business and negatively impact the trading price of our common shares.
Pursuant to Section 404 of the Sarbanes-Oxley Act, we will be required to furnish a report by our management on our internal control over financial reporting, and after we are no longer an emerging growth company, we will be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To achieve compliance with Section 404 within the prescribed period, we will have to engage in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. There is a risk that neither we nor our independent registered public accounting firm will be able to conclude within the prescribed timeframe that our internal control over financial reporting is effective as required by Section 404. This could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements.
Canadian laws differ from the laws in effect in the United States and may afford less protection to holders of our securities.
We are a Canadian corporation and are subject to the CBCA and applicable Canadian securities laws as a Canadian reporting issuer, which laws may differ from those governing a company formed under the laws of a United States jurisdiction. The provisions under the CBCA and other relevant laws may affect the rights of shareholders differently than those of a company governed by the laws of a United States jurisdiction, and may, together with our articles and by-laws, have the effect of delaying, deferring or discouraging another party from acquiring control of our company by means of a tender offer, a proxy contest or otherwise, or may affect the price an acquiring party would be willing to offer in such an instance.
We may be classified as a “passive foreign investment company,” which may have adverse U.S. federal income tax consequences for U.S. shareholders.
Generally, for any taxable year in which 75% or more of our gross income is passive income, or at least 50% of the average quarterly value of our assets (which may be determined in part by the market value of our common shares, which is subject to change) are held for the production of, or produce, passive income, we would be characterized as a passive foreign investment company (“PFIC”) for U.S. federal income tax purposes. Based on the price of our common shares and the composition of our gross assets (i) we believe that we were a PFIC for the taxable year ended December 31, 2016, (ii) we do not believe that we were a PFIC for the taxable year ended December 31, 2017 and (iii) we do not believe that we will be a PFIC for the taxable year ending December 31, 2018. Our status as a PFIC is a fact-intensive determination made on an annual basis, and we cannot provide any assurance regarding our PFIC status for the taxable year ending December 31, 2018 or for future taxable years.
If we are a PFIC for any year during a non-corporate U.S. shareholder’s holding period of our common shares, then such non-corporate U.S. shareholder generally will be required to treat any gain realized upon a disposition of our common shares, or any so-called “excess distribution” received on our common shares, as ordinary income, rather than as capital gain, and the preferential tax rate applicable to dividends received on our common shares would not be available. Interest charges would also be added to the taxes on gains and distributions realized by all U.S. holders.
A U.S. shareholder may avoid these adverse tax consequences by making a timely and effective “qualified electing fund” election (“QEF election”). A U.S. shareholder who makes a QEF election generally must report, on a current basis, its share of our ordinary earnings and net capital gains, whether or not we distribute any amounts to our shareholders. The QEF election is available only if the company characterized as a PFIC provides a U.S. shareholder with certain information regarding its earnings and capital gains as required under applicable U.S. Treasury regulations. In the event we become a PFIC, we intend to provide all information and documentation that a U.S. shareholder making a QEF election is required to obtain for U.S. federal income tax purposes (e.g., the U.S. shareholder’s pro rata share of ordinary income and net capital gain, and a “PFIC Annual Information Statement” as described in applicable U.S. Treasury regulations).
A U.S. shareholder may also mitigate the adverse tax consequences by timely making a mark-to-market election. A U.S. shareholder who makes the mark-to-market election generally must include as ordinary income each year the increase in the fair market value of the common shares and deduct from gross income the decrease in the value of such shares during each of its taxable years. A mark-to-market election may be made and maintained only if our common shares are regularly traded on a qualified exchange, including Nasdaq. Whether our common shares are regularly traded on a qualified exchange is an annual determination based on facts that, in part, are beyond our control. Accordingly, a U.S. shareholder might not be eligible to make a mark-to-market election to mitigate the adverse tax consequences if we are characterized as a PFIC.
Each U.S. shareholder should consult their own tax advisors with respect to the possibility of making these elections and the U.S. federal income tax consequences of the acquisition, ownership and disposition of our common shares. This paragraph is qualified in its entirety by the discussion in the section of this prospectus entitled “Certain United States Federal Income Tax Considerations.” In addition, our PFIC status may deter certain U.S. investors from purchasing our common shares, which could have an adverse impact on the market price of our common shares.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
Statements in this prospectus that are not descriptions of historical facts are forward-looking statements that are based on management’s current expectations and are subject to risks and uncertainties that could negatively affect our business, operating results, financial condition and share price. We have attempted to identify forward-looking statements by terminology including “anticipates,” “believes,” “can,” “continue,” “could,” “estimates,” “expects,” “intends,” “may,” “plans,” “potential,” “predicts,” “should,” “will,” “would” or the negative of these terms or other comparable terminology.
The forward-looking statements in this prospectus include, among other things, statements about:
|
● |
our plans to develop, obtain regulatory approval for and commercialize our DM199 product candidate for the treatment of AIS and CKD and our expectations regarding the benefits of our DM199 product candidate; |
|
● |
our ability to conduct successful clinical testing of our DM199 product candidate for AIS and CKD; |
|
● |
our ability to obtain required regulatory approvals of our DM199 product candidate for AIS and CKD; |
|
● |
the perceived benefits of our DM199 product candidate over existing treatment options for AIS and CKD; |
|
● |
the potential size of the markets for our DM199 product candidate and our ability to serve those markets; |
|
● |
the rate and degree of market acceptance, both in the United States and internationally, of our DM199 product candidate for AIS and CKD; |
|
● |
our ability to partner with and generate revenue from biopharmaceutical and pharmaceutical partners to develop, obtain regulatory approval for and commercialize our DM199 product candidate for AIS and CKD; |
|
● |
the success, cost and timing of planned clinical trials, as well as our reliance on collaboration with third parties to conduct our clinical trials; |
|
● |
our commercialization, marketing and manufacturing capabilities and strategy; |
|
● |
expectations regarding federal, state, and foreign regulatory requirements and developments, such as potential FDA regulation of our DM199 product candidate for AIS and CKD; |
|
● |
expectations regarding competition and our ability to obtain data exclusivity for our DM199 product candidate for acute ischemic stroke and chronic kidney disease; |
|
● |
our ability to obtain funding for our operations, including funding necessary to complete planned clinical trials and obtain regulatory approvals for our DM199 product candidate for AIS and CKD; |
|
● |
our estimates regarding expenses, future revenue, capital requirements and needs for additional financing; |
|
● |
our expectations regarding our ability to obtain and maintain intellectual property protection for our DM199 product candidate; |
|
● |
our expectations regarding the period during which we qualify as an emerging growth company under the JOBS Act; |
|
● |
the requirements of being a U.S. public reporting company; |
|
● |
our expectations regarding having our common shares listed on The Nasdaq Capital Market; and |
|
● |
our anticipated use of the net proceeds from this offering. |
These forward-looking statements are subject to a number of risks, uncertainties and assumptions, including those described under the heading “Risk Factors” in this prospectus Moreover, we operate in a very competitive and rapidly-changing environment. New risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. In light of these risks, uncertainties and assumptions, the forward-looking events and circumstances discussed in this prospectus may not occur and actual results could differ materially and adversely from those anticipated or implied in the forward-looking statements. You should not rely upon forward-looking statements as predictions of future events. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee that the future results, levels of activity, performance or events and circumstances reflected in the forward-looking statements will be achieved or occur. Except as required by law, including the securities laws of the United States, we do not intend to update any forward-looking statements to conform these statements to actual results or to changes in our expectations.
USE OF PROCEEDS
We estimate that our net proceeds from the sale of our common shares in this offering will be approximately $ million, or approximately $ million if the underwriter exercises its option in full to purchase additional shares from us, based on an assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, after deducting underwriting discounts and commissions and estimated offering expenses payable by us.
A $1.00 increase (decrease) in the assumed initial public offering price would increase (decrease) the net proceeds to us from this offering by approximately $ million, assuming the number of shares offered by us remains the same, and after deducting underwriting discounts and commissions and estimated offering expenses payable by us. Similarly, each increase or decrease of in the number of common shares offered by us would increase or decrease the net proceeds that we receive from this offering by approximately $ million, assuming that the assumed initial public offering price remains the same, and after deducting the underwriting discounts and commissions and estimated offering expenses payable by us.
We intend to use the net proceeds from this offering to fund clinical development of DM199, to conduct research activities and for working capital and general corporate purposes. We expect the net proceeds of this offering to be sufficient to allow us to complete our current Phase II Remedy trial in patients with acute ischemic stroke and a Phase 1b and Phase II study in patients with chronic kidney disease. We do not expect the net proceeds of this offering to be sufficient to fund, and we expect to require additional funding to complete, the development of DM199 through regulatory approval and commercialization, which we may seek through public or private equity or debt financings or through collaborations with other biotechnology companies or other sources. The expected use of the net proceeds from the offering represents our intentions based upon our current plans and business conditions. As of the date of this prospectus, we cannot predict with any certainty all of the particular uses for the net proceeds or the amounts that we will actually spend on the uses set forth above. The amounts and timing of our actual expenditures and the extent of product development and commercialization may vary significantly depending on numerous factors, including the status, results and timing of our planned clinical trials, as well as any unforeseen cash needs. As a result, our management will retain broad discretion over the allocation of the net proceeds from this offering.
Pending their use as described above, we plan to invest the net proceeds in short-term, interest-bearing obligations, investment-grade instruments, certificates of deposit or guaranteed obligations of the U.S. government. You will not have an opportunity to evaluate the economic, financial or other information on which we base our decisions regarding the use of these proceeds.
PRICE RANGE OF OUR COMMON SHARES
Our common shares trade in Canada on the TSX Venture Exchange under the trading symbol “DMA” and over-the-counter in the United States on the OTCQB marketplace under the trading symbol “DMCAF.” We have applied to list our common shares on The Nasdaq Capital Market under the trading symbol “DMAC.” The following tables set forth the quarterly high and low closing prices of our common shares on the TSX Venture Exchange and as quoted by the OTCQB for the fiscal quarters indicated. We have converted the trading prices on the TSX Venture Exchange to U.S. dollars using the exchange rate on the date of the corresponding high or low sales price. In quarters in which the high or low sales price occurred on multiple dates the exchange rate for the latest occurrence is used for purposes of converting the U.S. dollar amount. All prices in the table reflect the consummation of the 1-for-20 share consolidation of our common shares effected on November 15, 2018.
|
TSX Venture Exchange |
OTCQB |
|||||||||||||||||||||||
|
High |
High |
Low |
Low |
High |
Low |
|||||||||||||||||||
|
(CAD$) |
(US$) |
(CAD$) |
(US$) |
(US$) |
(US$) |
|||||||||||||||||||
|
Fiscal 2018 |
||||||||||||||||||||||||
|
Fourth Quarter (through November 16, 2018) |
$ | 11.60 | $ | 8.99 | $ | 6.10 | $ | 4.73 | $ | 9.20 | $ | 4.95 | ||||||||||||
|
Third Quarter |
17.60 | 13.40 | 9.40 | 7.20 | 13.40 | 7.20 | ||||||||||||||||||
|
Second Quarter |
16.00 | 12.40 | 7.60 | 5.80 | 12.20 | 4.00 | ||||||||||||||||||
|
First Quarter |
9.20 | 7.20 | 4.20 | 3.40 | 7.00 | 4.40 | ||||||||||||||||||
|
Fiscal 2017 |
||||||||||||||||||||||||
|
Fourth Quarter |
$ | 8.60 | $ | 6.80 | $ | 5.80 | $ | 4.60 | $ | 7.00 | $ | 3.80 | ||||||||||||
|
Third Quarter |
8.40 | 6.80 | 4.60 | 3.60 | 6.80 | 3.60 | ||||||||||||||||||
|
Second Quarter |
7.60 | 5.60 | 4.80 | 3.60 | 5.80 | 3.80 | ||||||||||||||||||
|
First Quarter |
5.40 | 4.00 | 2.80 | 2.20 | 4.20 | 2.20 | ||||||||||||||||||
|
Fiscal 2016 |
||||||||||||||||||||||||
|
Fourth Quarter |
$ | 4.80 | $ | 3.60 | $ | 3.20 | $ | 2.40 | $ | 4.00 | $ | 2.20 | ||||||||||||
|
Third Quarter |
6.80 | 5.20 | 4.20 | 3.20 | 5.20 | 3.60 | ||||||||||||||||||
|
Second Quarter |
6.60 | 5.20 | 2.80 | 2.20 | 5.00 | 3.20 | ||||||||||||||||||
|
First Quarter |
4.40 | 3.20 | 2.80 | 2.00 | 2.80 | 2.20 | ||||||||||||||||||
The last reported sale price for our common shares on the TSX Venture Exchange on November 16, 2018 was CAD $6.50 (US $4.94). The last reported sale price for our common shares as quoted by the OTCQB marketplace on November 16, 2018 was $4.95. As of November 15, 2018, we had 55 holders of record of our common shares. This does not include persons whose common shares are in nominee or “street name” accounts through brokers or other nominees.
DIVIDEND POLICY
We have never declared or paid cash dividends on our common shares, and currently do not have any plans to do so in the foreseeable future. We expect to retain our future earnings, if any, for use in the operation and expansion of our business. Moreover, we may in the future become subject to contractual restrictions on, or prohibitions against, the payment of dividends. Subject to the foregoing, the payment of cash dividends in the future, if any, will be at the discretion of our Board of Directors and will depend upon such factors as earnings levels, capital requirements, our overall financial condition and any other factors deemed relevant by our Board of Directors. As a result, you will likely need to sell your common shares to realize a return on your investment, and you may not be able to sell your shares at or above the price you paid for them.
CAPITALIZATION
The following table sets forth our cash and our capitalization as of September 30, 2018 on an actual basis and on an as adjusted basis to give effect to our issuance and sale of common shares at an assumed initial public offering price of $ per common share, which is the midpoint of the price range set forth on the cover page of this prospectus, after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us, and the application of the estimated net proceeds of this offering as described under “Use of Proceeds.” The information in this table is illustrative only and our capitalization following the completion of this offering will be adjusted based on the actual initial public offering price and other terms of this offering determined at pricing. You should read this table in conjunction with the information contained in “Use of Proceeds,” “Selected Financial Data” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” as well as the financial statements and the notes thereto included elsewhere in this prospectus.
|
As of September 30, 2018 |
||||||||
|
Actual |
As Adjusted(1) |
|||||||
|
(unaudited) (in thousands) |
||||||||
|
Cash |
$ | 3,898 | ||||||
|
Shareholders’ equity |
||||||||
|
Common shares, no par value, unlimited authorized, actual and as adjusted, 7,839,176 shares issued and outstanding, actual; and shares issued and outstanding, as adjusted |
— | |||||||
|
Additional paid-in capital |
48,116 | |||||||
|
Accumulated deficit |
(44,006 | ) | ||||||
|
Total shareholders’ equity |
4,110 | |||||||
|
Noncontrolling interest |
— | |||||||
|
Total shareholders’ equity |
4,110 | |||||||
|
Total capitalization |
4,110 | |||||||
__________________________
(1) Each $1.00 increase (decrease) in the assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, would increase (decrease) each of cash, additional paid-in capital, total shareholders’ equity and total capitalization by approximately $ million, assuming the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting underwriting discounts and commissions and estimated offering expenses payable by us. We may also increase or decrease the number of shares we are offering. Each increase (decrease) of shares in the number of shares offered by us would increase (decrease) each of cash, additional paid-in capital, total shareholders’ equity and total capitalization by approximately $ million, assuming that the assumed initial public offering price, which is the midpoint of the price range set forth on the cover page of this prospectus, remains the same, and after deducting underwriting discounts and commissions and estimated offering expenses payable by us. The as adjusted information discussed above is illustrative only and will adjust based on the actual initial public offering price and other terms of this offering determined at pricing.
The number of common shares issued and outstanding as set forth in the table above excludes:
|
● |
639,359 common shares issuable upon the exercise of stock options outstanding under the DiaMedica Therapeutics Inc. Stock Option Plan as of September 30, 2018, at a weighted-average exercise price of $7.87 per share; |
|
● |
21,183 common shares issuable upon the settlement of deferred share units outstanding the DiaMedica Therapeutics Inc. Deferred Share Unit Plan as of September 30, 2018; |
|
● |
123,374 common shares reserved for future issuance under the DiaMedica Therapeutics Inc. Stock Option and DiaMedica Therapeutics Inc. Deferred Share Unit Plans as of September 30, 2018; |
|
● |
825,264 common shares issuable upon the exercise of outstanding warrants as of September 30, 2018, at a weighted-average exercise price of $6.67 per share; and |
|
● |
common shares issuable upon the exercise of the warrant that will be issued to the underwriter in connection with this offering, with an exercise price equal to 120% of the initial public offering price per share. |
DILUTION
Purchasers of common shares in this offering will experience immediate dilution to the extent of the difference between the initial public offering price per share of our common shares and the net tangible book value per share of common share immediately after this offering.
Our net tangible book value as of September 30, 2018 was approximately $4.1 million, or $0.52 per common share. Net tangible book value per share is determined by dividing the net of total tangible assets less total liabilities, by the aggregate number of common shares outstanding as of September 30, 2018. After giving effect to the sale by us of common shares at an assumed initial public offering price of $ per common share, which is the midpoint of the price range set forth on the cover page of this prospectus, and after deducting the underwriting discounts and commissions and estimated offering expenses, our net tangible book value as of September 30, 2018 would have been approximately $ million, or $ per common share. This represents an immediate increase in net tangible book value of $ per share to our existing shareholders and an immediate dilution of $ per common share issued to the new investors purchasing common shares in this offering.
The following table illustrates this per share dilution to new investors:
|
Assumed initial public offering price per common share |
$ | |||||||
|
Net tangible book value per share as of September 30, 2018 |
$ | 0.52 | ||||||
|
Increase in net tangible book value per share attributable to this offering |
$ | |||||||
|
Net tangible book value per share after this offering |
$ | |||||||
|
Dilution per share to new investors participating in this offering |
$ |
Each $1.00 increase or decrease in the assumed initial public offering price of $ per common share, which is the midpoint of the price range set forth on the cover page of this prospectus, would increase or decrease the dilution per common share to new investors purchasing common shares in this offering by $ per share, assuming that the number of common shares offered by us, as set forth on the cover page of this prospectus, remains the same, and after deducting underwriting discounts and commissions and estimated offering expenses payable by us.
We may also increase or decrease the number of common shares we are offering. An increase of shares in the number of common shares we are offering would increase our as adjusted net tangible book value by $ per share and decrease the dilution to new investors in this offering by $ per share, assuming that the assumed initial public offering price remains the same, and after deducting the estimated underwriting discounts and commissions and the estimated offering expenses payable by us. Similarly, a decrease of shares in the number of common shares we are offering would decrease our as adjusted net tangible book value by $ per share and increase the dilution to new investors in this offering by $ per share, assuming that the assumed initial public offering price remains the same, and after deducting the underwriting discounts and commissions and estimated offering expenses.
If the underwriter exercises its option to purchase additional common shares in full in this offering at the assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, the net tangible book value per share after this offering would be $ per share, the increase in the net tangible book value per share to existing shareholders would be $ per share and the dilution to new investors purchasing securities in this offering would be $ per share.
The above table excludes:
|
● |
639,359 common shares issuable upon the exercise of stock options outstanding under the DiaMedica Therapeutics Inc. Stock Option Plan as of September 30, 2018, at a weighted-average exercise price of $7.87 per share; |
|
● |
123,374 common shares reserved for future issuance under the DiaMedica Therapeutics Inc. Stock Option and DiaMedica Therapeutics Inc. Deferred Share Unit Plans as of September 30, 2018; |
|
● |
21,183 common shares issuable upon the settlement of deferred share units under our Deferred Share Unit Plan as of September 30, 2018; |
|
● |
825,264 common shares issuable upon the exercise of outstanding warrants as of September 30, 2018, at a weighted-average exercise price of $6.67 per share; and |
|
● |
common shares issuable upon the exercise of the warrant that will be issued to the underwriter in connection with this offering, with an exercise price equal to 120% of the initial public offering price per share. |
To the extent that options or warrants are exercised, new options are issued under our stock option plan, or we issue additional common shares in the future, there may be further dilution to investors participating in this offering. Moreover, we may choose to raise additional capital because of market conditions or strategic considerations, even if we believe that we have sufficient funds for our current or future operating plans. If we raise additional capital through the sale of equity or convertible debt securities, the issuance of these securities could result in further dilution to our shareholders.
SELECTED FINANCIAL DATA
The following tables present, as of the dates and for the periods indicated, our selected historical financial data and certain as adjusted financial data, as indicated therein. The consolidated statements of operations data for the years ended December 31, 2016 and 2017 and the consolidated balance sheet data as of December 31, 2016 and 2017 are derived from our audited financial statements that are included elsewhere in this prospectus. The summary consolidated statements of operations data for the nine months ended September 30, 2017 and 2018 and the consolidated balance sheet data as of September 30, 2018 are derived from the unaudited condensed consolidated financial statements included elsewhere in this prospectus. We have prepared the unaudited interim consolidated financial statements on the same basis as the audited consolidated financial statements and have included, in our opinion, all adjustments, consisting only of normal recurring adjustments, that we consider necessary for a fair statement of financial statements set forth in those statements. Our historical results are not indicative of the results to be expected in the future and our interim results are not necessarily indicative of results to be expected for the full year ending December 31, 2018, or any other period.
You should read this information together with our financial statements and the related notes, as well as the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this prospectus.
|
Year Ended |
Nine Months |
|||||||||||||||
|
2016 |
2017 |
2017 |
2018 |
|||||||||||||
|
(unaudited) |
||||||||||||||||
|
(in thousands, exept share and per share data) |
||||||||||||||||
|
Consolidated Statements of Operations Data: |
||||||||||||||||
|
Operating revenues: |
||||||||||||||||
|
License revenue |
$ | - | $ | - | $ | - | $ | 500 | ||||||||
|
Operating expenses: |
||||||||||||||||
|
Research and development |
1,728 | 3,206 | 2,577 | 3,071 | ||||||||||||
|
General and administrative |
598 | 1,313 | 941 | 2,073 | ||||||||||||
|
Total operating expenses |
2,326 | 4,519 | 3,518 | 5,144 | ||||||||||||
|
Loss from operations |
(2,326 | ) | (4,519 | ) | (3,518 | ) | (4,644 | ) | ||||||||
|
Other (income) expense |
||||||||||||||||
|
Governmental assistance - research incentives |
- | (244 | ) | (244 | ) | (1,046 | ) | |||||||||
|
Other (income) expense |
82 | (6 | ) | 14 | 61 | |||||||||||
|
Change in fair value of warrant liability |
(188 | ) | (9 | ) | 208 | 39 | ||||||||||
|
Total other (income) expense |
(106 | ) | (259 | ) | (22 | ) | (946 | ) | ||||||||
|
Loss before income tax |
(2,220 | ) | (4,260 | ) | (3,496 | ) | (3,698 | ) | ||||||||
|
Income tax |
- | - | - | 74 | ||||||||||||
|
Net loss & comprehensive loss |
$ | (2,220 | ) | $ | (4,260 | ) | $ | (3,496 | ) | $ | (3,772 | ) | ||||
|
Loss per share, basic and diluted |
$ | (0.47 | ) | $ | (0.72 | ) | $ | (0.60 | ) | $ | (0.51 | ) | ||||
|
Weighted average number of shares outstanding: |
||||||||||||||||
|
Basic and diluted |
4,735,751 | 5,935,790 | 5,848,178 | 7,406,378 | ||||||||||||
| Consolidated Balance Sheet Data: | ||||||||||||||||
|
As of December 31, |
As of |
|||||||||||||||
|
2016 |
2017 |
September 30, 2018 |
||||||||||||||
|
Actual |
Actual |
Actual |
As Adjusted |
|||||||||||||
|
(unaudited) |
||||||||||||||||
|
Cash |
$ | 1,736 | $ | 1,353 | $ | 3,898 | $ | |||||||||
|
Working capital |
1,092 | 491 | 3,748 | |||||||||||||
|
Total assets |
1,875 | 1,802 | 5,463 | |||||||||||||
|
Total current liabilities |
764 | 1,003 | 1,353 | |||||||||||||
|
Total stockholders' equity |
1,111 | 799 | 4,110 | |||||||||||||
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following Management’s Discussion and Analysis of Financial Condition and Results of Operations is based upon accounting principles generally accepted in the United States of America and discusses the financial condition and results of operations for DiaMedica Therapeutics Inc. for the three and nine months ended September 30, 2018 and 2017 and the years ended December 31, 2017 and 2016.
This discussion should be read in conjunction with our consolidated financial statements and related notes included elsewhere in this prospectus. The following discussion contains forward-looking statements that involve numerous risks and uncertainties. Our actual results could differ materially from the forward-looking statements as a result of these risks and uncertainties. See “Special Note Regarding Forward-Looking Statements” for additional cautionary information.
Overview
We are a clinical stage biopharmaceutical company primarily focused on the development of novel recombinant proteins. Our goal is to use our patented and licensed technologies to establish our company as a leader in the development and commercialization of therapeutic treatments for novel recombinant proteins to treat neurological and kidney diseases. Our current primary focus is on AIS and CKD. We plan to advance DM199, our lead drug candidate, through required clinical trials to create shareholder value by establishing its clinical and commercial potential as a therapy for AIS and CKD.
We have not generated any revenues from product sales. Since our inception, we have financed our operations from public and private sales of equity, the exercise of warrants and stock options, interest income on funds available for investment, and government grants and tax credits. We have incurred losses in each year since our inception. Our net losses were $3.8 million and $3.5 million for the nine months ended September 30, 2018 and 2017, respectively, and $4.3 million and $2.2 million for the years ended December 31, 2017 and 2016, respectively. As of September 30, 2018, we had an accumulated deficit of $44.0 million. Substantially all of our operating losses resulted from expenses incurred in connection with product candidate development programs, our R&D activities and G&A support costs associated with our operations.
We expect to continue to incur significant expenses and increasing operating losses for at least the next several years. In the near term, we anticipate that our expenses will increase as we:
|
● |
advance the ongoing clinical development of DM199; |
|
● |
maintain, expand and protect our intellectual property portfolio; and |
|
● |
provide G&A support for our operations. |
To fund future operations we will need to raise additional capital. The amount and timing of future funding requirements will depend on many factors, including the timing and results of our ongoing development efforts, the potential expansion of our current development programs, potential new development programs and related G&A support. We anticipate that we will seek to fund our operations through public or private equity or debt financings or other sources, such as potential collaboration agreements. We cannot assure you that anticipated additional financing will be available to us on favorable terms, or at all. Although we have previously been successful in obtaining financing through our equity securities offerings, there can be no assurance that we will be able to do so in the future.
Financial Overview
Revenues
Since our inception, we have incurred losses while advancing the R&D of our therapeutic product candidates. We have not generated any revenues from product sales and do not expect to do so for a number of years. We may never generate revenues from our DM199 product candidate or any of our preclinical development programs, as we may never succeed in obtaining regulatory approval or commercializing any of these product candidates.
Research and Development Expenses
R&D expenses consist primarily of fees paid to external service providers such as contract research organizations and contract manufacturing organizations related to clinical trials, contractual obligations for clinical development, clinical sites, laboratory testing, preclinical trials, development of DM199 and the related manufacturing processes, salaries, benefits, share-based compensation and other personnel costs. We spent $3.1 million and $2.6 million on R&D expenses for the nine months ended September 30, 2018 and 2017, respectively, and $3.2 million and $1.7 million for the years ended December 31, 2017 and 2016, respectively. Over the past approximately eight years, our R&D efforts have been primarily focused on DM199 for AIS and CKD.
At this time, due to the risks inherent in the clinical development process and the early stage of our product development programs, we are unable to estimate with any certainty the costs we will incur in the continued development of DM199 or any of our preclinical development programs. We expect that our R&D expenses may increase if we are successful in advancing DM199, or any of our preclinical programs, into advanced stages of clinical development. The process of conducting clinical trials necessary to obtain regulatory approval and manufacturing scale-up to support expanded development and potential future commercialization is costly and time consuming. Any failure by us or delay in completing clinical trials, manufacturing scale-up or in obtaining regulatory approvals could lead to increased R&D expense and, in turn, have a material adverse effect on our results of operations.
General and Administrative Expenses
G&A expenses consist primarily of salaries and related benefits, including share-based compensation related to our executive, finance, business development and support functions. Other G&A expenses include rent and utilities, travel expenses and professional fees for auditing, tax and legal services. We expect that G&A expenses will increase in the future as we expand our operating activities. In addition, G&A expenses are expected to reflect increased costs associated with our anticipated U.S. public reporting company status and listing on The Nasdaq Capital Market. We anticipate incurring one-time costs associated with this offering of approximately $400,000 in 2018, consisting primarily of the Nasdaq listing process and legal and accounting fees.
Other (Income) Expense
Other (income) expense consists primarily of governmental assistance – research incentives, change in the fair value of our warrants that are accounted for as derivative liabilities, interest income, and foreign currency exchange gains and losses. In 2016, other expense was partially offset by the $250,000 gain recognized from the sale of a previous technology no longer being developed by the Company.
Critical Accounting Policies and Estimates
Management’s discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles. The preparation of these consolidated financial statements requires us to make estimates and assumptions for the reported amounts of assets, liabilities, revenue, expenses and related disclosures. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions and any such differences may be material.
While our significant accounting policies are more fully described in Note 4 to our consolidated financial statements included elsewhere in this prospectus, we believe the following discussion addresses our most critical accounting policies, which are those that are most important to the portrayal of our financial condition and results of operations and require our most difficult, subjective and complex judgments.
Revenue recognition
We followed ASC 606, “Revenue from Contracts with Customers” in accounting for our License and Collaboration agreement with Ahon Pharma. Accordingly, the Company recognizes revenue upon transfer of control of the product to our customer in an amount that reflects the consideration we expect to receive in exchange.
We intend to enter into arrangements for the research and development (R&D) and/or manufacture of products and product candidates. Such arrangements may require us to deliver various rights, services and/or goods, including (i) intellectual property rights or licenses, (ii) R&D services or (iii) manufacturing services. The underlying terms of these arrangements generally would provide for consideration to DiaMedica in the form of nonrefundable, up-front license fees, development and commercial-performance milestone payments, cost sharing, royalty payments and/or profit sharing.
In arrangements involving more than one performance obligation, each required performance obligation is evaluated to determine whether it qualifies as a distinct performance obligation based on whether (i) the customer can benefit from the good or service either on its own or together with other resources that are readily available and (ii) the good or service is separately identifiable from other promises in the contract. The consideration under the arrangement is then allocated to each separate distinct performance obligation based on its respective relative stand-alone selling price. The estimated selling price of each deliverable reflects our best estimate of what the selling price would be if the deliverable was regularly sold by us on a stand-alone basis or using an adjusted market assessment approach if selling price on a stand-alone basis is not available.
The consideration allocated to each distinct performance obligation is recognized as revenue when control of the related goods or services is transferred. Consideration associated with at-risk substantive performance milestones is recognized as revenue when it is probable that a significant reversal of the cumulative revenue recognized will not occur. We intend to utilize the sales and usage-based royalty exception in arrangements that resulted from the license of intellectual property, recognizing revenues generated from royalties or profit sharing as the underlying sales occur.
Share-based Compensation
We account for all share-based compensation awards using a fair value method. The cost of employee and non-employee services received in exchange for awards of equity instruments is measured and recognized based on the estimated grant date fair value of those awards. Compensation cost is recognized ratably using the straight-line attribution method over the vesting period, which is considered to be the requisite service period. We record forfeitures in the periods in which they occur.
The fair value of share-based awards is estimated using the Black-Scholes option pricing model. The determination of the fair value of share-based awards is affected by our stock price, as well as assumptions regarding a number of complex and subjective variables. Risk free interest rates are based upon Canadian Government bond rates appropriate for the expected term of each award. Expected volatility rates are based on the on historical volatility equal to the expected life of the option. The assumed dividend yield is zero, as we do not expect to declare any dividends in the foreseeable future. The expected term of options is estimated considering the vesting period at the grant date, the life of the option and the average length of time similar grants have remained outstanding in the past.
The assumptions used in calculating the fair value under the Black-Scholes option valuation model are set forth in the following table for options issued by the Company for the years ended December 31, 2017 and 2016:
|
2017 |
2016 |
||
|
Common share fair value |
$0.26 - $0.42 |
$0.16 - $0.24 |
|
|
Risk-free interest rate |
1.1% |
0.8% |
|
|
Expected dividend yield |
0% |
0% |
|
|
Expected option life |
4.5 years |
4.6 years |
|
|
Expected stock price volatility |
84.7 – 156.8% |
92.0 – 185.1% |
Recently Issued Accounting Pronouncements
In February 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2016-02, Leases. The guidance in ASU 2016-02 supersedes the lease recognition requirements in the Accounting Standards Codification Topic 840, Leases. ASU 2016-02 requires an entity to recognize assets and liabilities arising from a lease for both financing and operating leases, along with additional qualitative and quantitative disclosures. The new standard requires the immediate recognition of all excess tax benefits and deficiencies in the income statement and requires classification of excess tax benefits as an operating activity as opposed to a financing activity in the statements of cash flows. ASU 2016-02 is effective for fiscal years beginning after December 15, 2018, with early adoption permitted. Management is evaluating the impact of the new standard on our consolidated financial statements.
In June 2018, the FASB issued ASU 2018-07, Compensation – Stock Compensation (Topic 718): Improvements to Nonemployee Share-Based Payment Accounting, which simplifies several aspects of the accounting for nonemployee share-based payment transactions resulting from expanding the scope of Topic 718 to include share-based payment transactions for acquiring goods and services from nonemployees. This ASU is effective for the Company for fiscal years beginning after December 15, 2018, including interim periods within that fiscal year. Early adoption is permitted, but no earlier than an entity’s adoption date of Topic 606. Management is currently evaluating the impact of the new guidance on our consolidated financial statements.
Recently adopted accounting pronouncements
In May 2014, the Financial Accounting Standards Board (FASB) issued a new accounting standard that amends the guidance for the recognition of revenue from contracts with customers to transfer goods and services. The FASB subsequently issued additional, clarifying standards to address issues arising from implementation of the new revenue recognition standard. The new revenue recognition standard and clarifying standards require an entity to recognize revenue when control of promised goods or services is transferred to the customer at an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. We adopted this new standard as of January 1, 2018, but the adoption as of this date had no impact on our financial statements, as we had no revenue until the third quarter of 2018.
Results of Operations
Comparison of the Three and Nine Months Ended September 30, 2018 and 2017
The following table summarizes our results of operations for the three and nine months ended September 30, 2018 and 2017. The table below summarizes our revenue and expenses (in thousands):
|
Three Months Ended September 30, |
Nine Months Ended September 30, |
|||||||||||||||
|
2018 |
2017 |
2018 |
2017 |
|||||||||||||
|
License revenue |
$ | 500 |
|
$ | — | $ | 500 |
|
$ | — | ||||||
|
Research and development |
1,210 | 411 | 3,071 | 2,577 | ||||||||||||
|
General and administrative |
777 | 415 | 2,073 | 941 | ||||||||||||
|
Other (income) |
(157 |
) |
(119 |
) |
(946 |
) |
(22 | ) | ||||||||
License Revenue
License revenue is the current year is comprised of the initial $500,000 license payment we were entitled to receive upon signing of the September 27, 2018, license and collaboration agreement with Ahon Pharma.
Research and Development Expenses
R&D expenses were $1.2 million and $411,000 for the three months ended September 30, 2018 and 2017, respectively. R&D expenses were $3.1 million for the nine months ended September 30, 2018, an increase of approximately $494,000 from $2.6 million in the same period of 2017. These increases over the comparable prior year periods were due primarily to additional pre-clinical testing and related costs required to support an application for an investigational new drug application in the United States, higher study costs for the REMEDY Phase 2 stroke study as compared with the DM199 bridging study which was in progress during the comparable prior year period and increased non-cash stock-based compensation costs. These increases were partially offset by reduced patent prosecution and employee recruiting costs.
General and Administrative Expenses
G&A expenses were $777,000 for the three months ended September 30, 2018 compared to $415,000 for the same period in 2017. G&A expenses were $2.1 million for the first nine months of 2018 compared to $941,000 for the same period in 2017. G&A expenses increased in both periods due to one-time costs incurred associated with our anticipated U.S. public offering, primarily the Nasdaq listing process and legal and accounting fees, and increased salaries, fees and short-term benefits due to the addition of staff. Share-based compensation expense increased related to the recognition of expense for awards granted during 2017 and 2018.
Other (Income) Expense
Other income was $157,000 for the three months ended September 30, 2018 compared to $119,000 for the same period in 2017. Other income was $946,000 for the nine months ended September 30, 2018 compared to $22,000 for the same period in 2017. These increases in other income resulted primarily from the recognition of the R&D incentive from the Australian government for qualifying research work performed by DiaMedica Australia and by reduced charges recorded for the change in fair value of derivative warrant liability related to common share purchase warrants which expired in February 2018. These increases were partially offset by increased foreign currency transaction losses.
Comparison of the Years Ended December 31, 2017 and 2016
The following table summarizes our results of operations for the years ended December 31, 2017 and 2016 (in thousands):
|
Year Ended December 31, |
||||||||
|
2017 |
2016 |
|||||||
|
Research and development |
$ | 3,206 | $ | 1,728 | ||||
|
General and administrative |
1,313 | 598 | ||||||
|
Other (income) expense |
(259 | ) | (106 | ) | ||||
Research and Development Expenses
R&D expenses were $3.2 million for the year ended December 31, 2017 compared to $1.7 million for the year ended December 31, 2016, an increase of $1.5 million. The increase is primarily due to the costs incurred in conjunction with the advancement of the DM199 clinical trial program. Salaries, fees, and short-term benefits and share-based compensation also increased for the year ended December 31, 2017 over the comparable prior year period due to an increase in staff to support the clinical program.
General and Administrative Expenses
G&A expenses were $1.3 million for the year ended December 31, 2017 compared to $598,000 for the year ended December 31, 2016. General and administrative costs increased slightly due to an increase in outsourced services and salaries, fees, and short-term benefits, which were mainly due to an increase in staff. These increases were partially offset by decreased share-based compensation resulting from a reduction in the number of grants during 2017.
Other (Income) Expense
Other (income) expense was $259,000 in income for the year ended December 31, 2017 compared to $106,000 in income for 2016. Other income for 2017 increased due to the recognition of government assistance in the form of the R&D incentive tax credit received from Australia, related to qualifying clinical trial and other research expenses incurred by our Australian subsidiary.
Liquidity, Capital Resources and Going Concern
Since our inception, we have incurred losses while advancing the R&D of our therapeutic product candidates. We have not generated any revenues from product sales and do not expect to do so for a number of years. We do not know when, or if, we will generate any revenue from our product candidates. We do not expect to generate any revenue from sales of our product candidates unless and until we obtain regulatory approval. At the same time, we expect our expenses to increase in connection with our ongoing development activities, particularly as we continue the research, development and clinical trials of, and seek regulatory approval for, our product candidates. In addition, we expect to incur additional costs associated with operating as a U.S. public reporting company. In addition, subject to obtaining regulatory approval of any of our product candidates, we expect to incur significant commercialization expenses for product sales, marketing, manufacturing and distribution. We anticipate that we will need additional funding in connection with our continuing operations.
Since our inception, we have financed our operations from public and private sales of equity, the exercise of warrants and stock options, interest income on funds available for investment, and government grants and tax credits. We had cash totaling $3.9 million and $1.4 million and working capital of $3.7 million and $491,000 as of September 30, 2018 and December 31, 2017, respectively.
On March 29, 2018, we completed, in two tranches, a brokered and non-brokered private placement of 1,322,965 units at a price of $4.90 per unit for aggregate gross proceeds of approximately $6.3 million. Each unit consisted of one common share and one-half of one common share purchase warrant. Each whole warrant entitles the holder to purchase one common share at a price of $7.00 at any time prior to expiration on March 19, 2020 and March 29, 2020 for tranche 1 and tranche 2, respectively. The warrants are subject to early expiration under certain conditions. In connection with the offering, we paid an aggregate cash fee of approximately $384,000 to brokers and issued an aggregate of 80,510 compensation options. Each compensation option entitles the holder to purchase one common share at $4.90, the offering price, for a period of two years from the closing of the offering, subject to acceleration on the same terms as the warrants issued to the investors.
The report of our independent registered public accounting firm on our December 31, 2017 audited consolidated financial statements includes an explanatory paragraph referring to our ability to continue as a going concern. In the next 12 months, we will likely seek to raise additional funds through various sources, such as equity and debt financings, or through strategic collaborations and license agreements. This additional funding will be required to continue our R&D and other operating activities as we have not reached successful commercialization of our product candidates. These circumstances cast significant doubt as to our ability to continue as a going concern. Our consolidated financial statements have been prepared assuming that we will continue as a going concern. Our future operations are expected to continue to be dependent upon our ability to secure additional funds, negotiate license agreements with partners and/or generate product revenues in order to fully execute our business plan. There can be no assurance that we will be successful in commercializing our products, entering into strategic agreements with partners, raising additional capital on favorable terms or that these or other strategies will be sufficient to permit us to continue as a going concern.
The amount and timing of future funding requirements will depend on many factors, including the timing and results of our ongoing development efforts, the potential expansion of our current development programs, potential new development programs and related G&A support. We anticipate that we will seek to fund our operations through public or private equity or debt financings or other sources, such as potential collaboration agreements. We cannot assure you that anticipated additional financing will be available to us on favorable terms, or at all. Although we have previously been successful in obtaining financing through our equity securities offerings, there can be no assurance that we will be able to do so in the future. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interests of our shareholders will be diluted. Debt financing, if available, may involve agreements that include conversion discounts or covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through government or other third-party funding, marketing and distribution arrangements or other collaborations, or strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us.
Cash Flows
Operating Activities
Cash used in operating activities for the nine months ended September 30, 2018 was $3.8 million compared to $2.9 million for the nine months ended September 30, 2017. This decrease relates primarily to a reduction in the net loss, partially offset by the effects of the changes in operating assets and liabilities.
Cash used in operating activities for the year ended December 31, 2017 was $3.9 million, compared to $3.0 million for the year ended December 31, 2016, an increase of $0.9 million. This increase relates primarily to the increase in net loss, partially offset by an increase in non-cash charges for share-based compensation and the effects of changes in operating assets and liabilities.
Investing Activities
Investing activities consist primarily of purchases of property and equipment. Net cash used in investing activities was $63,000 for the nine months ended September 30, 2018 compared to $7,000 for the nine months ended September 30, 2017 and was $22,000 for the year ended December 31, 2017 compared to $7,000 for the year ended December 31, 2016.
Financing Activities
Financing activities consist primarily of net proceeds from the sale of common shares and warrants and proceeds from the exercise of stock options and warrants. Net cash provided by financing activities was $6.4 million for the nine months ended September 30, 2018 compared to $2.0 million for the nine months ended September 30, 2017.
Net cash provided by financing activities was $3.5 million for the year ended December 31, 2017 compared to $4.6 million for the year ended December 31, 2016, a decrease of $1.1 million.
Cash flows from financing activities included net proceeds from the following private placements of our common shares and warrants to purchase common shares:
|
● |
On March 29, 2018, we completed, in two tranches, a brokered and non-brokered private placement of 1,322,965 units at a price of $4.90 per unit for aggregate gross proceeds of approximately $6.3 million. Each unit consisted of one common share and one-half of one common share purchase warrant. Each whole warrant entitles the holder to purchase one common share at a price of $7.00 at any time prior to expiration on March 19, 2020 and March 29, 2020 for tranche 1 and tranche 2, respectively. The warrants are subject to early expiration under certain conditions. In connection with the offering, we paid an aggregate cash fee of approximately $384,000 to brokers and issued an aggregate of 80,510 compensation options. Each compensation option entitles the holder to purchase one common share at $4.90, the offering price, for a period of two years from the closing of the offering, subject to acceleration on the same terms as the warrants issued to the investors. |
|
● |
On December 18, 2017, we completed a non-brokered private placement of 181,220 units at a price of $5.20 per unit for aggregate gross proceeds of approximately $944,000. Each unit consisted of one common share and one-half of one common share purchase warrant. Each whole warrant entitles the holder to purchase one common share at a price of $7.00 at any time prior to expiration on December 19, 2019, subject to early expiration under certain conditions. |
|
● |
On April 17, 2017, we completed a non-brokered private placement of 526,316 units at a price of $3.80 per unit for aggregate proceeds of approximately $2,000,000. Each unit consists of one common share and one-half common share purchase warrant. Each whole warrant entitles the holder to purchase one common share at a price of $4.60 at any time prior to expiration on April 17, 2019. The warrant expiration date can be accelerated at our option in the event that the volume-weighted average trading price of our common shares exceeds $6.00 per common share for any 10 consecutive trading days. |
|
● |
On September 8, 2016, we completed the second tranche of a non-brokered private placement of 750,000 common shares at a price of $4.00 per share for aggregate gross proceeds of $3,000,000. |
|
● |
On August 22, 2016, we completed the first tranche of a non-brokered private placement of 250,000 common shares at a price of $4.00 per share for aggregate gross proceeds of $1,000,000. |
|
● |
On February 25, 2016, we completed the second tranche of a non-brokered private placement of 43,750 units at a price of $2.34 per unit for aggregate gross proceeds of approximately $101,710. Each unit consisted of one common share and one-half of one common share purchase warrant. Each whole warrant entitled the holder to purchase one common share at a price of CAD$5.00 at any time prior to expiration of two years from the closing date. |
|
● |
On February 18, 2016, we completed the first tranche of a non-brokered private placement of 190,625 units at a price of $2.34 per unit for aggregate gross proceeds of approximately $445,544. Each unit consisted of one common share and one-half of one common share purchase warrant. Each whole warrant entitled the holder to purchase one common share at a price of CAD$5.00 at any time prior to expiration of two years from the closing date. |
While our rate of future negative cash flow per month will vary due to the timing of expenses incurred, at the current rate of negative cash flow per month we believe that our current cash will enable us to complete our currently ongoing Phase II trial in patients with AIS and initiate a Phase Ib trial in patients with CKD. Our future cash requirements will increase if we decide to expand our R&D efforts beyond the currently planned development of DM199.
Commitments and Contingencies
In the normal course of business, we incur obligations to make future payments as we execute our business plan. As of September 30, 2018, we had outstanding commitments, including R&D contracts and other commitments, that are known and committed of approximately $1.9 million over the next 12 months and approximately $400,000 in the following 12 months. These contracts relate to preclinical, clinical, and development activities, including the clinical research organization conducting the Phase II clinical trial for DM199 related to AIS. These commitments are subject to significant change and the ultimate amounts due may be materially different as these obligations are affected by, among other factors, the number and pace of patients enrolled, the number of clinical study sites, amount of time to complete study enrollments and the time required to finalize the analysis and reporting of study results. These commitments are generally cancelable upon 30 days’ notice, with our obligation then limited to costs incurred up to that date. As of September 30, 2018, we had future operating lease commitments totaling approximately $240,000 over the remainder of the lease, of which $62,000 is due over the next 12 months.
We have entered into a license agreement with Catalent Pharma Solutions, LLC (“Catalent”) whereby we have licensed certain gene expression technology and we contract with Catalent for the manufacture of DM199. Under the terms of this license, certain milestone and royalty payments may become due under this agreement and are dependent upon, among other factors, clinical trials, regulatory approvals and ultimately the successful development of a new drug, the outcome and timing of which is uncertain. As of September 30, 2018, two milestones remain which include $185,000 due upon the initiation of dosing in our first Phase III trial and $185,000 upon our first regulatory approval for commercial sale. Following the launch of our first product, we will also incur a royalty of less than 1% on net sales. The royalty term is indefinite but may be canceled by us on 90 days’ prior written notice. The license may not be terminated by Catalent unless we fail to make required milestone and royalty payments.
Off-Balance Sheet Arrangements
During 2017 and 2016 and the nine months ended September 30, 2018, we did not have any off-balance sheet arrangements (as defined by applicable SEC regulations) that are reasonably likely to have a current or future material effect on our financial condition, results of operations, liquidity, capital expenditures or capital resources.
Internal Control Over Financial Reporting
Pursuant to Section 404(a) of the Sarbanes-Oxley Act, commencing the year following our first annual report required to be filed with the SEC, our management will be required to report on the effectiveness of our internal control over financial reporting. The rules governing the standards that must be met for management to assess our internal control over financial reporting are complex and require significant documentation, testing and possible remediation. To comply with the requirements of being a reporting company under the Exchange Act, we may need to upgrade our systems, including information technology, implement additional financial and management controls, reporting systems and procedures and hire additional accounting and finance staff.
BUSINESS
Overview
We are a clinical stage biopharmaceutical company primarily focused on the development of novel recombinant (synthetic) proteins. Our goal is to use our patented and licensed technologies to establish our company as a leader in the development and commercialization of novel recombinant proteins to treat neurological and kidney diseases. Our primary focus is on acute ischemic stroke (“AIS”) and chronic kidney disease (“CKD”). We plan to advance our lead drug candidate, DM199, through clinical trials, as appropriate, to create shareholder value by establishing its clinical and commercial potential as a therapy for AIS and CKD.
DM199 is a recombinant form of human tissue kallikrein-1 (“KLK1”). KLK1 is a serine protease (protein) produced in the pancreas, kidneys and salivary glands, which plays a critical role in the regulation of local blood flow and vasodilation (the widening of blood vessels which decreases blood pressure) in the body, as well as an important role in inflammation and oxidative stress (an imbalance between potentially damaging reactive oxygen species, or free radicals, and antioxidants in your body). We believe DM199 has the potential to treat a variety of diseases where healthy functioning requires sufficient activity of KLK1 and its system, the kallikrein-kinin system (“KKS”). The primary focus for our DM199 program development is on AIS and CKD; however, we also intend to pursue advancement in the vascular dementia market.
The current status of our product candidates in preclinical and clinical development is as follows:
KLK1 is involved in multiple biochemical processes. The most well-characterized activity of KLK1 is its enzymatic cleavage of low molecular weight kininogen (“LMWK”) to produce bradykinin (“BK”)-like peptides, collectively known as kinins, which activate BK receptors (BK1R, BK2R). Activation of BK receptors by kinins sets in motion metabolic pathways that can improve blood flow (through vasodilation), dampen inflammation, and protect tissues and end-organs from ischemic damage. Scientific literature, including publications in Circulation Research, Immunopharmacology and Kidney International, suggests that lower endogenous KLK1 levels in patients are associated with diseases related to vascular disorders, such as kidney diseases, stroke and hypertension. We believe DM199 could replenish endogenous KLK1 to properly activate the BK system that protects the kidney and brain from damage. By providing this additional supply of KLK1, DM199 treatment could improve blood flow to damaged end-organs, such as kidneys and brain, supporting the structural integrity and normal functioning.
DM199 (KLK1): Increasing Blood Flow in Brain and Kidneys
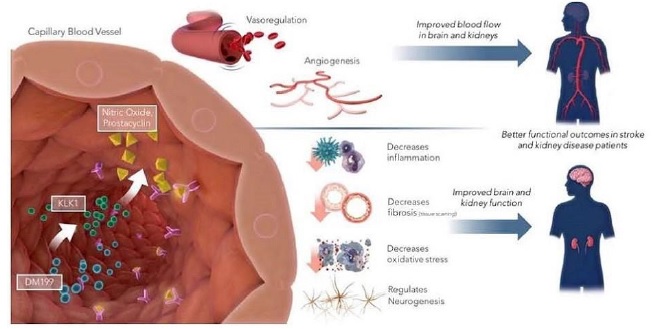
We believe DM199 may provide new treatment options with significant benefits over the current standards of care by offering potentially fewer side effects and a therapeutic treatment option to a greater number of patients. There are no approved therapies in the United States or the European Union, of which we are aware, to address low KLK1 levels. We are positioning DM199 for worldwide use. We have conducted and are conducting clinical trials in Europe and Australia to support regulatory filings in the United States, Europe and around the world; with an initial focus on the United States. We are currently preparing to file an initial IND application with the FDA in the United States in patients with CKD.
Lower KLK1 levels are associated with initial stroke events and are also a predictor of stroke recurrence after an initial stroke. As shown in the graph below, the red line represents patients in the lowest KLK1 quartile who are at the highest risk for recurrence of stroke. (2,478 stroke patients and event free survival over 5 years).
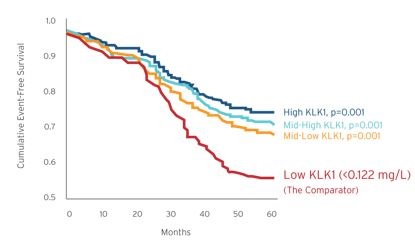
For patients suffering from kidney disease, studies have shown that KLK1 excretion, or levels of KLK1 in the urine, significantly decreased in patients with mild kidney disease and was further reduced in patients with severe renal failure requiring dialysis as compared to healthy subjects, as illustrated in the graph below.
Low KLK1 Levels Associated With Kidney Disease
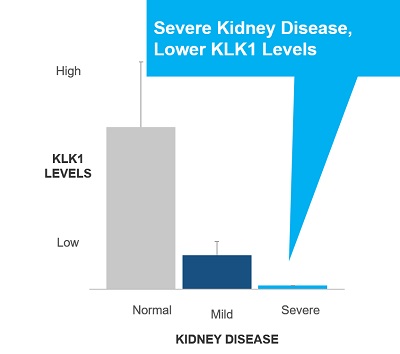
Our Strategy
Our goal is to become a leader in the discovery, development, and commercialization of recombinant proteins for the treatment of severe and life-threatening diseases. We seek to identify and select, for development and partnership, recombinant proteins with novel mechanisms that have biological properties with broad applicability. Once we have selected a class of recombinant proteins, we apply their biological properties to clinical settings with unmet needs, and we evaluate opportunities based on estimated development timelines and costs, regulatory pathway, and commercial opportunities. After identifying suitable molecules for clinical development, we intend to mitigate development risk by maintaining a diversified and broad clinical pipeline, rapidly analyzing data to determine the potential of each program and entering into development collaborations with industry-leading companies.
Currently, our strategy includes the following key components:
|
● |
DM199 for AIS - complete our ongoing Phase II study |
|
● |
DM199 for CKD - advance Phase Ib and Phase II studies |
|
● |
DM199 for vascular dementia - initiate Phase II study, following AIS study and with sufficient resources |
|
● |
Leverage our technologies to expand our development pipeline |
|
● |
Use our expertise to identify and manufacture novel recombinant proteins |
Targeted Indications and Markets for DM199
Acute Ischemic Stroke
Stroke is characterized by the rapidly developing loss of brain function due to disturbance in the blood. As a result, the affected area of the brain becomes inactive and eventually dies. Strokes can be classified into two major categories: AIS and hemorrhagic stroke. AIS is characterized by interruption of the blood supply by a blood clot (ischemia), while a hemorrhagic stroke results from rupture, or bleeding, of a blood vessel or an abnormal vascular structure. According to the U.S. Center for Disease Control and Prevention (“CDC”), about 87% of strokes are ischemic in nature with the remainder classified as hemorrhagic. According to the CDC, worldwide, stroke is an important cause of adult disability and the second leading cause of death in developed countries. Risk factors for stroke include advanced age, hypertension (high blood pressure), previous stroke or transient ischemic attack (“TIA”), diabetes, high cholesterol, cigarette smoking and atrial fibrillation. According to the World Health Organization, each year approximately 15 million people worldwide suffer a stroke, of which 5.5 million will die and 5.0 million will be permanently disabled. According to the CDC:
|
● |
Every year in the United States, approximately 795,000 people experience a new or recurrent stroke each year (ischemic or hemorrhagic). Approximately 610,000 of these are first events and 185,000 are recurrent stroke events. |
|
● |
Stroke caused approximately one of every 20 deaths in the United States. On average, someone in the United States has a stroke every 40 seconds, and someone dies from a stroke every four minutes. |
|
● |
Stroke costs the United States $34 billion annually, including the cost of health care services, medications and lost productivity. |
At the site of a blood flow blockage in the brain, there exist two major ischemic zones - the core ischemic zone with nearly complete loss of blood flow, and the surrounding ischemic penumbra having partially reduced blood flow. Within minutes, the significant lack of blood flow in the core (i.e. glucose and oxygen deprivation) rapidly depletes energy stores and triggers the loss of ion gradients, ultimately leading to neuronal cell death. The ischemic penumbra zone, however, may remain viable for several hours via collateral arteries that branch from the main occluded artery in the core zone. Unfortunately, the penumbra is at great risk of delayed tissue damage due to inflammation and cell death, or apoptosis. As time goes on, a lack of blood flow in the ischemic zone (infarct) leads to fluid buildup (edema) and swelling which creates intracranial pressure. This pressure on the brain leads to tissue compression resulting in additional ischemia. Additional events in AIS include vascular damage to the blood vessel lining or endothelium, loss of structural integrity of brain tissue and blood vessels, and inflammation. A stroke can lead to permanent damage with memory loss, speech problems, reading and comprehension difficulties, physical disabilities, and emotional/behavioral problems. The long-term costs of stroke are substantial, with many patients requiring extended hospitalization, extended physical therapy or rehabilitation, and/or long-term institutional or family care. However, provided the extended window of viability in the penumbra, next generation stroke therapies are being developed to protect valuable brain tissue during the hours to a week after a stroke.
Acute Ischemic Stroke Treatment Options
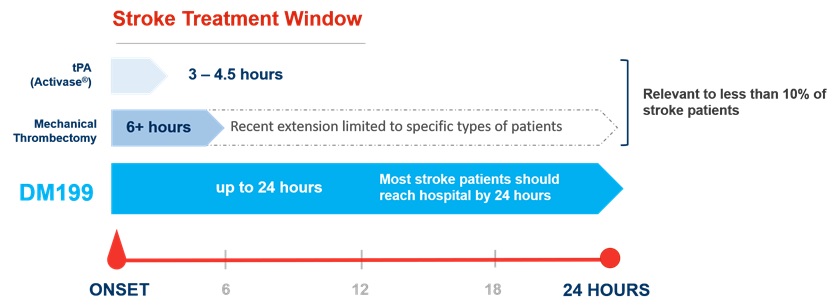
We believe that stroke represents an area of significant unmet medical need, and a KLK1 treatment (such as DM199) could provide a treatment option and a significant patient benefit with its proposed therapeutic window of up to 24 hours after the first sign of symptoms. Currently, the only pharmacological intervention for AIS is the use of tissue plasminogen activator (“tPA”), which must be given within 4.5 hours of symptom onset. Mechanical thrombectomy, in which the clot is removed using catheter-based tools, is also available to some patients. Despite the availability of these treatments, many patients are not eligible due to the location of the clot, the elapsed time after the stroke occurred, or safety considerations. Thus, we believe DM199 offers significant advantages over the current treatment options and fills an unmet need for patients who cannot receive tPA. Additionally, DM199 may also offer a complimentary follow-on treatment for patients who initially receive tPA or mechanical thrombectomy treatments. Based on the number of strokes each year (approximately 1.7 million in the U.S., Europe and Japan and 15 million worldwide) and the $8,500 estimated cost per patient for the current standard of care, tPA, we believe the annual market opportunity for DM199 could be significant.
DM199 Acute Ischemic Stroke: Proposed Mechanism
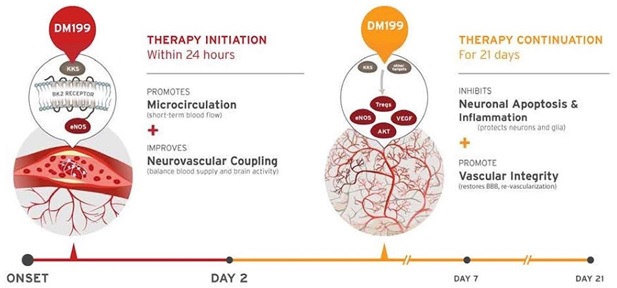
KLK1 in China (marketed under the brand name Kailikang®) is widely used for the treatment of AIS, making therapy available to hundreds of thousands of patients who currently have no options. Kailikang® is a human urine-extracted KLKl protein. We believe that the proprietary DM199 protein could result in an improved efficacy with optimized pharmacokinetics (drug level exposure) and avoid the side effects of risk of endotoxins, impurities and antibody formation in comparison to Kailikang® that is isolated from human urine. We also believe that DM199 addresses potential supply constraints that makes Kailikang® difficult and expensive to produce given the limited source of human urine. We believe these factors make the recombinant protein DM199 a product candidate that is better positioned for regulatory approval worldwide than a urine-derived protein since we believe it can meet the rigorous required manufacturing standards.
Chronic Kidney Disease
CKD is characterized by a progressive decline in overall kidney function as measured by glomerular filtration rate (“GFR”) (a test used to check how well the kidneys are filtering excess fluid and waste products out of your blood), and albuminuria (the amount of albumin protein excreted in your urine). When GFR gets too low, patients develop end stage renal disease (“ESRD”) and require dialysis or a kidney transplant to survive. Among multiple underlying causes, CKD often begins with an increase in blood glucose, which leads to the thickening of the glomerular membrane, known as fibrosis. As the kidney function becomes impaired, GFR decreases and abnormal amounts of protein are released into the urine collecting tubules of the kidney through damaged capillary pores. Additionally, increased blood glucose leads to increased blood pressure, reactive oxygen species, advanced glycation end product formation (harmful compounds that are formed when protein or fat combine with sugar in the bloodstream) and inflammation. As this continues, structural components of the kidney (the nephron) begin to collapse, resulting in cell ischemia and cell death. As the renal damage continues, a progressive thickening of the basement membrane is seen along with continued pathological changes in the cell and inflammation. Early stages of CKD are characterized as microalbuminuria (small amounts of protein leak into the urine). Late stages are characterized as macroalbuminuria (large amount of protein in the urine). The rate of decline depends on the type of diabetes, genetic predisposition, glycemic controls, and blood pressure. At the final stages of CKD, the kidneys fail completely and dialysis or a kidney transplant is needed.
CKD is a widespread health problem that generates significant economic burden throughout the world, including:
|
● |
30 million Americans and 120 million Chinese suffer from this debilitating and potentially life-threatening condition according to the National Kidney Foundation. |
|
● |
The primary causes of CKD are diabetes (Type 2 and Type 1) and hypertension. The Medical Clinics of North America estimates that over 40% of those with Type 2 diabetes and 20% of those with Type 1 diabetes will eventually develop CKD, making it one of the more common risks for diabetics. |
|
● |
Patients with CKD are at greater risk for hypertension and heart disease. |
Currently, there is no cure for CKD and treatment involves management of the disease. Blood pressure medications, such as angiotensin converting enzyme inhibitors (“ACEi”) or angiotensin receptor blockers (“ARB”), are often prescribed to control hypertension, and hopefully, slow the progression of CKD. Nevertheless, according to the National Kidney Foundation, many patients continue to show declining kidney function, with the overall population having a lifetime risk of 3.6% of developing ESRD, where dialysis or a kidney transplant are needed. We believe DM199 offers a potentially novel approach for the treatment CKD since KLK1 protein plays a vital role in normal kidney function, and BK and BK receptors are critical for kidney health and integrity. Since patients with moderate to severe CKD often excrete abnormally low levels of KLK1 in their urine, we believe that DM199 may prevent or reduce further kidney damage by replenishing endogenous KLK1 and restoring the protective BK system.
Potential Treatments with DM199
Acute Ischemic Stroke
We believe treatment of AIS with DM199 could have both immediate and long-term benefits for patients that could significantly improve outcomes following AIS. Immediate actions include activation of the KKS to release nitric oxide and improve microcirculation in ischemic tissue along with improvements in the balance between blood flow and brain activity (neurovascular coupling). Long-term (days following the stroke) actions include the restoration of the blood brain barrier through increases in T regulatory cells (“T-regs” – a subpopulation of T cells that modulate the immune system and prevent autoimmune disease) and inhibition of apoptotic cell death.
In China, a human urine-extracted KLKl protein (Kailikang®) is approved and marketed by Techpool Bio-Pharma Inc., a company controlled by Shanghai Pharmaceuticals Holding Co. Ltd. We believe Kailikang® has been approved for the treatment of AIS in China with a treatment window of up to 48 hours post-stroke. Based on IQVIA data, other publications and internal estimates, we believe over 500,000 stroke patients have been treated with Kailikang® for acute ischemic stroke in Asia. More than 50 published clinical studies, covering over 4,000 stroke patients, have demonstrated a beneficial effect of Kailikang® treatment in AIS. According to a publication in the China Journal of Neurology, in a double-blinded, placebo-controlled trial of 446 patients treated with either KLK1 or a placebo administered up to 48 hours after a stroke showed significantly better scores on the European Stroke Scale and Activities of Daily Living at three weeks post-treatment and after three months using the Barthel Index.
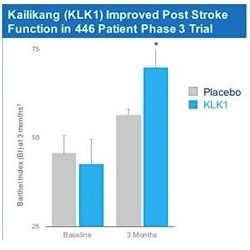
Furthermore, a comprehensive meta-analysis covering 24 clinical studies involving 2,433 patients published in the Journal of Evidenced Based Medicine concluded that human urinary KLK1 appears to ameliorate neurological deficits for patients with AIS and improves long-term outcomes, though a few treated patients suffered from transient hypotension.
As DM199 is a recombinant form of human KLK1, we believe it has the potential to preserve “at risk” brain tissue by increasing cerebral blood flow, establishing better collateral circulation, decreasing inflammation, reducing cell death, or apoptosis, and facilitating improved blood flow to at-risk ischemic penumbra brain tissue. We believe DM199 offers the potential for an improved recombinant product for worldwide use. We are developing DM199 to treat AIS patients with therapy beyond the current window of 3 to 4.5 hours for tPA to up to 24 hours after the first sign of symptoms, thereby filling a large unmet need of patients who cannot receive tPA under the currently available treatment window of tPA. We believe this could potentially make therapy available to the millions of patients worldwide who currently have limited options.
Chronic Kidney Disease
We also believe DM199 has the potential to offer therapeutic benefits for CKD patients. The KLK1 protein plays a vital role in normal kidney function, and BK and BK receptors are critical for kidney health and integrity. Patients with moderate to severe CKD often excrete abnormally low levels of KLK1 in their urine, leading to the hypothesis that this KLK1 deficit contributes to disease progression. We believe that DM199 may replenish endogenous KLK1 and activate the BK system that protects the kidney from damage. In fact, DM199 treatment in an animal model of Type 1 diabetes delayed the onset of the disease, attenuated the degree of insulitis (inflammation in the insulin producing islet cells of the pancreas) and improved pancreatic beta cell mass in a dose-dependent manner by increasing T-regs. By providing additional KLK1, DM199 has the following potentially beneficial actions:
|
● |
Improve blood flow to the kidney by restoring proper regulation of blood flow through veins arteries and especially capillaries (vasoregulation); |
|
● |
Support the structural integrity of the kidney by reducing scar tissue formation (fibrosis), oxidative stress, and inflammation; and |
|
● |
Activate mechanisms that upregulate T-regs, improve insulin sensitization, glucose uptake and glycogen synthesis, and lower blood pressure. |
Further supporting the hypothesis that an intact KKS is critical for normal kidney function, a series of observational studies published in Immunopharmacology showed the amount of KLK1 released into the urine appears to be inversely correlated with the severity of disease in patients with CKD. Urinary KLK1 excretion was decreased in patients with both mild (not requiring dialysis) and severe (kidney failure/hemodialysis) renal disease compared to controls. The severity of the disease was negatively correlated with KLK1 excretion. Decreases in urinary KLK1 activity was seen especially when the reduction was associated with decreased glomerular filtration rate. We believe DM199 may potentially have advantages over ACEi because it restores already depleted KLK1 levels.
DM199 treatment is intended to directly replenish KLK1 levels, normalizing kidney function. Current treatment options, especially ACEi drugs, only partially restore kidney function and are associated with high-risk side effects. Importantly, it is becoming increasingly clear that part of the beneficial effect of ACEi drugs involves preventing the normal breakdown of BK leading to substantial increases in BK levels throughout the body. While higher BK levels benefit the kidney, ACEi drugs can generate excessive BK where is it not needed, potentially leading to side effects such as persistent cough, angioedema (swelling of skin and tissue) and hyperkalemia (abnormally high potassium levels that can lead to cardiac arrest and sudden death). We believe DM199 treatment could allow KLK1 to follow its normal physiological processes and release BK when and where it is needed, avoiding these side effects. Importantly, we believe successful treatment with ACEi in kidney disease requires a fully functional kallikrein kinin system, KLK1 and bradykinin systems, potentially making ACEi drugs less effective in patients with a pre-existing KLK1 deficit.
KLK1 derived from the pancreas of a pig, or porcine KLK1, is currently used to treat CKD in China and Japan. Porcine KLK1 is also used to treat hypertension and retinopathy in Japan, China and Korea. Based on IQVIA data and our estimates, we estimate millions of patients have been treated with porcine KLK1 for CKD, retinopathy and other vascular diseases in Asia. Over 20 clinical papers have been published in the Chinese literature supporting the therapeutic activity in CKD patients of porcine KLK1 given alone or in combination with an ARB or an ACEi. These unblinded studies involve treatment durations ranging from a few weeks up to six months and report improvement in kidney disease based on decreased urinary albumin excretion rates and other clinical endpoints of kidney disease.
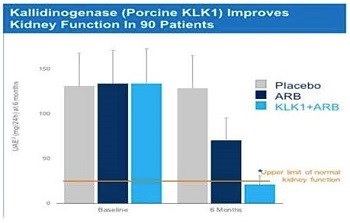
There is a significant need for new and alternative treatment strategies for CKD and we believe that the combined results of these studies, which are consistent with our proposed mechanism of action for and preclinical studies of DM199, provide a good rationale for formal clinical development of DM199. We intend to seek approval for worldwide use of DM199 as a novel and ground-breaking therapy for CKD. We believe DM199 could potentially complement the use of ACEi or ARBs to improve kidney functions without increasing the risk for hyperkalemia, chronic cough, angiodema or other related side effects. Less than 30% of patients with CKD are believed to be on optimal dose of ACEi or ARB due in part to risk of hyperkalemia which can lead to cardiac arrest and sudden death. We believe DM199, through the activation of the BK system, may complement the renin-angiotensin system, primarily targeted by ACEi and ARBs. Activation of the BK system may improve the function of the diseased renal system by improving vasodilation and insulin sensitization, as well as blocking fibrosis, inflammation, thrombosis and oxidative stress. A significant potential advantage of DM199 over ACEi/ARB treatments is that hyperkalemia may be less likely with DM199. We anticipate that DM199 will boost KLK1 levels to release physiological levels of BK when and where needed, generating beneficial nitric oxide and prostacyclin while increasing regulatory T cells (T-regs or TREGS) to reduce inflammation. In addition, porcine KLK1 has demonstrated the ability to directly cleave vascular endothelial growth factor (“VEGF”) in laboratory tests using vitreous fluid extracted from human eyes. This may contribute to the efficacy of porcine KLK1 reported in patients with diabetic macular edema. Porcine KLK1 is currently marketed in Japan for this indication.
DM199 (Recombinant KLK1), ACEi, ARB and Plasma Kallikrein Proposed Mechanism of Actions
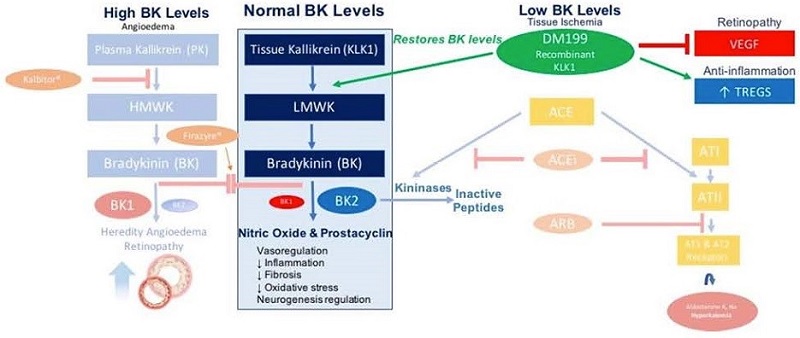
Other Potential Programs
We are also currently developing a diagnostic tool, DMDx, to measure KLK1 levels. Several published studies indicate KLK1 insufficiency is associated with multiple disease states including hypertension, CKD and AIS. Levels of endogenous KLK1 in both urine and plasma are inversely correlated with disease severity. Importantly, the decrease in urinary protein occurs in a disease state (e.g. CKD), where a primary hallmark is increased secretion of many other proteins. In this way, we believe KLK1 is a potentially unique diagnostic tool for such diseases.
We believe DM199 may also offer a potentially novel treatment for vascular dementia patients. Vascular dementia is caused by chronic impaired blood supply within the brain, often associated with TIA or prior stroke. According to the Alzheimer’s Society, one third of all stroke survivors could develop dementia within five years. According to the US National Institute of Neurological Disorders and Stroke, there are over 6 million stroke survivors in the U.S. alone. In a clinical study, KLK1 isolated from human urine demonstrated the ability to improve cognitive function in vascular dementia patients and increase cerebral blood flow. We have drafted a protocol synopsis for a Phase II study in vascular dementia. Our decision to commence this study will be dependent upon our cash resources and efficacy data from our other DM199 studies.
Our Competition and Current Treatments for Acute Ischemic Stroke and Chronic Kidney Disease
The biopharmaceutical industry is highly competitive and characterized by rapidly advancing technologies that focus on rapid development of proprietary drugs. We believe that our product candidates, development capabilities, experience and scientific knowledge provide us with competitive advantages. However, we face significant potential competition from many different sources, including major pharmaceutical, specialty pharmaceutical and biotechnology companies, academic institutions, governmental agencies and other research institutions. Any product candidates that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future.
Many of our competitors, either alone or with their strategic partners, have substantially greater financial, technical and human resources than we do, and experience in obtaining U.S. Food and Drug Administration (“FDA”) and other regulatory approvals of treatments and commercializing those treatments. Accordingly, our competitors may be more successful than us in obtaining approval for competitive products and achieving widespread market acceptance. Our competitors’ treatments may be more effectively marketed and sold than any products we may commercialize, thus limiting our market share and resulting in a longer period before we can recover the expenses of developing and commercializing our product candidates.
Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These activities may lead to consolidated efforts that allow for more rapid development of competitive product candidates.
We also compete for staff, development and clinical resources. These competitors may impair our ability to recruit or retain qualified scientific and management personnel, our ability to work with specific advisors, clinical contract organizations, due to conflicts of interest or capacity constraints, and may also delay recruitment of clinical study sites and study volunteers, impeding progress in our development programs.
We expect any products that we develop and commercialize to compete on the basis of, among other things, efficacy, safety, price and the availability of reimbursement from government or other third-party payors. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are viewed as safer, more effective or less expensive than any products that we may develop.
Acute Ischemic Stroke
Currently, there is one approved pharmaceutical treatment for acute ischemic stroke. That treatment is tPA (marketed under the brand name Activase®), and its therapeutic window is limited to 3 to 4.5-hours after the AIS. There are, however, a number of companies that are actively pursuing a variety of approaches to develop pharmaceutical products for the treatment of AIS including, among others:
|
● |
Stem cells (Athersys, Inc.) |
|
● |
Cerebral edema (Biogen Inc.) |
|
● |
Anti-inflammatory and clot dissolving (Biogen Inc.) |
|
● |
Cell protection and anti-inflammation (ZZ Biotech LLC) |
|
● |
Inhibits platelet aggregation (Acticor Biotech SAS) |
We believe that there is a large unmet therapeutic need for AIS treatments that can be administered beyond the 3 to 4.5-hour time window of tPA. With this large unmet therapeutic need, there is significant competition to develop new therapeutic options. New therapeutic options in development include tissue protection focused therapies (deliverable from hours to days after the stroke) that preserve and protect brain cells beyond the tPA therapeutic window. Currently, the most advanced treatments involve the mechanical removal of blood clots in brain arteries through sophisticated catheter-based approaches. According to published research, use of mechanical thrombectomy is growing and the window of time after a stroke where the procedure can be used is widening. These therapies are especially targeted toward preserving viable cells in the ischemic penumbra hours after a stroke. The goal is to provide treatment options for the vast majority of AIS patients who do not receive hospital care early enough to qualify for tPA therapy. We believe there is a very significant market opportunity for a drug that has a therapeutic window beyond that of tPA and is able to obtain regulatory approval.
Chronic Kidney Disease
In the United States, we are aware of only one currently approved treatment for CKD. That treatment is an ACEi (marketed under the brand name Captopril®) which is approved for the treatment of patients with CKD caused by Type 1 diabetes. There are several pharmaceutical products for the treatment of CKD currently in clinical development, some of which include:
|
● |
Mineralcorticortisteroid receptor agonist (Bayer HealthCare Pharmaceuticals LLC) |
|
● |
CCR2 receptor antagonists (ChemoCentryx, Inc., Bristol-Myers Squibb Company) |
|
● |
Oxidative stress, cyclo-oxygenase 2 inhibitors (Reata Pharmaceuticals, Inc.) |
|
● |
Glycosylation inhibitors (Glycadia, Inc. aka Glycadia Pharmaceuticals) |
|
● |
Endothelin A receptor antagonists (AbbVie Inc.) |
|
● |
Cyclin nucleotide phosphodiesterase inhibitor (Pfizer Inc.) |
|
● |
Aldosterone receptor antagonists (Mitsubishi Tanabe Pharma Corporation) |
|
● |
Nitric oxide enzyme inhibitor (GenKyoTex SA) |
|
● |
Nitric oxide (Ironwood Pharmaceuticals, Inc.) |
Current treatment strategies for CKD include the strict control of high blood pressure and high blood sugar. The ACEi drug Captopril is approved for use in patients with CKD due to Type 1 diabetes and both ACEi and ARBs are widely prescribed to slow the progression of CKD. However, according to the National Kidney Foundation, 3.6% of the U.S. population over their lifetime will develop ESRD requiring dialysis or kidney transplantation. Furthermore, the treatment with ACEi and ARBs has been linked to hyperkalemia (elevated blood potassium levels), which increases the risk for abnormal heart rhythms and sudden death. In fact, two clinical trials investigating the use of ACEi and ARB combination therapy in kidney disease were stopped prematurely because participants developed hyperkalemia. The added complication of hyperkalemia results in patients receiving suboptimal dosing or patients being untreated because they cannot tolerate the treatment. Additional side effects with ACEi treatment are angioedema (swelling of skin tissue) and persistent cough.
DM199 treatment is intended to directly replenish KLK1 levels, normalizing kidney function. Current treatment options, especially ACEi drugs, only partially restore kidney function and the association with high-risk side effects. ACEi drugs can generate excessive BK where is it not needed, potentially leading to related side effects such as cough and angioedema (swelling of skin and tissue). We believe DM199 treatment would potentially allow KLK1 to follow its normal physiological processes and release BK when and where it is needed, avoiding these side effects. Importantly, successful treatment with ACEi in kidney disease requires a fully functional KLK1 system, potentially making ACEi drugs less effective in patients with a pre-existing KLK1 deficit.
DM199 Clinical Studies
We have completed five clinical trials with DM199 in over 120 volunteers, including multiple Phase I single dose ascending and multiple dose ascending studies in healthy volunteers and patients with Type 2 diabetes. Chronic dosing studies over 16 to 28 days were also conducted in healthy volunteers and patients with Type 2 diabetes. (see Table 1 below). As is generally the case for early phase clinical trials, the primary endpoints for all studies were safety, tolerability, and pharmacokinetics. The Phase II (Part D) study also investigated a series of secondary endpoints that included blood glucose concentration, insulin levels, glucose tolerance testing and a variety of experimental biomarkers of evaluating the potential efficacy of DM199 in treating Type 2 diabetes patients.
Table 1 DM199 Trial Design Overview
|
Trial |
Participants (N) |
Design |
Doses (µg/kg) |
Route |
Length |
|
Phase-I Part A |
Healthy (32) |
Single ascending dose |
5, 15, 30, 50 |
SC |
1 week |
|
Phase-I Part B |
Type 2 diabetes (10) |
Single ascending dose |
0.3, 1.5, 15 |
SC |
1 week |
|
Phase-I Part C |
Healthy (18) |
Multiple ascending dose |
3, 15, 25 |
SC |
6 doses over 16 days |
|
Phase-IIA Part D |
Type 2 diabetes (36) |
Blinded multiple dose |
Placebo, 3, 15 |
SC |
10 doses over 28 days |
|
Phase I Bridging |
Healthy (36) |
Single ascending dose |
0.25, 0.50. 0.75, 1.0 3.0 |
IV IV SC |
1 week |
In combination, these studies showed that DM199 was well tolerated and demonstrated clear physiological activity. After subcutaneous (“SC”) injection (under the skin), DM199 exhibited a favorable pharmacokinetic profile with extended half-life (i.e., the time required to reduce concentration of the drug in the body by one-half), supporting potential dosing intervals of up to one week. The dose-limiting tolerability issue in healthy volunteers was orthostatic hypotension (a condition in which blood pressure falls significantly when a person stands) observed largely at the 50 µg/kg dose level, which is much greater than those anticipated to be efficacious in patients. In each trial, observed treatment emergent side-effects were mild to moderate in severity and resolved completely. The most common treatment-emergent side effects included headache, dizziness, nausea and injection site pain, the majority of which were observed in the highest dose group of the Phase I-Part A trial.
Two of our clinical studies have focused on patients with Type 2 diabetes. The first study enrolled 10 Type 2 diabetic patients. The patients were dosed with either DM199, at three single ascending dose levels or placebo. DM199 was well-tolerated at all three dose levels by the diabetic patients with no dose limiting side effects. The second study in patients with Type 2 diabetes enrolled 36 patients treated with one of two subcutaneous dose levels of DM199 or placebo over 28 days. This study achieved its primary endpoints and demonstrated that DM199 was well-tolerated. The secondary endpoints for this study, however, were not met. The secondary efficacy endpoints were confounded due to what we believe were significant execution errors caused by protocol deviations occurring at the clinical trial site that were unable to be reconciled. See “Business—Legal Proceedings” for more information on this study.
In February 2018, we initiated treatment on the first patient in our Phase II REMEDY trial assessing the safety, tolerability and markers of therapeutic efficacy of DM199 in patients suffering from AIS. Our REMEDY trial is expected to enroll 60 patients to evaluate DM199 in patients with AIS. We intend to use a portion of the proceeds from this offering to expand enrollment to 90-100 patients. The study drug (DM199 or placebo) will be administered as an intravenous (“IV”) infusion within 24 hours of stroke symptom onset, followed by SC injections once every 3 days for 21 days. The study is designed to measure safety and tolerability along with multiple tests designed to investigate DM199’s therapeutic potential including plasma-based biomarkers and standard functional stroke measures assessed at 90 days post-stroke. Standard functional stroke measurements include the Modified Rankin Scale, National Institutes of Health Stroke Scale, the Barthel Index and C-reactive protein, a measure of inflammation.
In March 2018, we had an in-person meeting with the Office of Drug Evaluation, Cardiovascular and Renal Division, of the FDA. The purpose of the meeting was to gain feedback and recommendations from the FDA on our planned clinical study of DM199 in patients with CKD. The study endpoints are expected to include:
|
● |
identifying dose(s) of DM199 that may normalize plasma concentrations of KLK1; |
|
● |
demonstrating safety and tolerability; and |
|
● |
evaluating standard measures of kidney function and treatment biomarkers. |
Based on the FDA’s guidance, we expect the study to include patients suffering from mild to moderate CKD (stage 3) due to Type 1 and Type 2 diabetes and will be designed to test multiple dosing strategies. Standard measures of safety, DM199 plasma levels and kidney function will be collected before, during and after DM199 treatment. We intend to file an IND application for this study in the fourth quarter of 2018. This study is intended to help identify the proper dosing strategy for future efficacy trials of DM199 for CKD.
In 2017, we completed and published in the International Journal of Clinical Trials the results from a Phase lb study with DM199 designed to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics in healthy volunteers. Specifically, this study compared multiple doses levels of DM199, administered via IV and subcutaneous routes to identify a dose and delivery route that most closely compared to or improves upon the pharmacokinetic and pharmacodynamics profile of the approved urinary KLK1 in China. We found that a dose of DM199 administered via IV infusion mimicked the drug profile of IV-administered urinary derived KLK1 (Kailikang®). We believe that this study also identified a dose of DM199, administered via subcutaneous injection, which had a superior pharmacokinetic profile and that maintained more normal KLK1 levels throughout day. Below are results from our clinical trial showing the pharmacokinetic profile of subcutaneously administered DM199 observed in study subjects as compared to what we believe is normal range in healthy subjects.
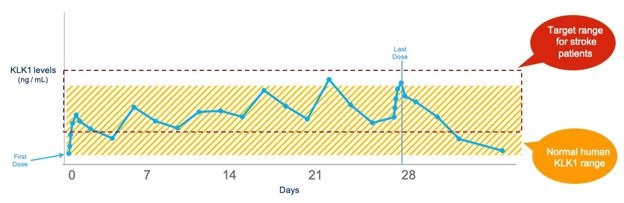
Potential DM199 Commercial Advantages
Several researchers have studied the structural and functional properties of KLK1. This deep body of knowledge has revealed the potential clinical benefits of KLK1 treatments. Today, forms of KLK1 derived from human urine and porcine pancreas are sold in Japan, China and Korea to treat acute ischemic stroke, chronic kidney disease, retinopathy, hypertension and related diseases. We are not aware of any synthetic version of KLK1 with regulatory approval for human use in any country, nor are we aware of any synthetic version in development besides our drug candidate DM199 (recombinant human KLK1). We believe at least five companies have attempted to create a synthetic version of KLK1.
The growing understanding of KLK1’s role in human health and its use in Asia as an approved therapeutic highlight two important potential commercial advantages for DM199:
|
● |
KLK1 treatment is sold in Japan, China and Korea. Research has shown that patients with low levels of KLK1 are associated with a variety of diseases related to vascular dysfunction, such as chronic kidney disease, acute ischemic strokes, retinopathy and hypertension. Clinical trial data with human urine and porcine KLK1 has demonstrated statistically significant clinical benefits of treating a variety of patients with KLK1 compared to placebo. These efficacy results are further substantiated by established markets in Japan, China and Korea for pharmaceutical sales of KLK1 derived from human urine and porcine pancreas. |
|
● |
KLK1 treatment has had limited side effects and has been well tolerated in studies to date. KLK1 is naturally produced by the human body; and therefore, the body’s own control mechanisms act to limit potential side effects. The only notable side effect observed in our clinical trials was orthostatic hypotension, or sudden drop in blood pressure, which was only seen at doses significantly higher than our anticipated therapeutic dose levels. Routine clinical use of KLK1 treatment in Asia has been well-tolerated by patients. In 2017, we completed a clinical trial comparing the pharmacokinetic profile of DM199 to Kailikang® for acute ischemic stroke, which showed DM199, when administered in intravenous form, to have a profile similar to Kailikang®. Further, when DM199 was administered subcutaneously, DM199 demonstrated a superior, longer acting, pharmacokinetic profile to Kailikang®. |
We have conducted numerous internal and third-party analyses to demonstrate that DM199 is structurally and functionally equivalent to KLK1 derived from human urine. The amino acid structure of DM199 is identical to the human urine form, and the enzymatic and pharmacokinetic profiles are substantially similar to both human urinary and porcine derived KLK1. The physiological effects of DM199 on blood pressure, from our completed studies, mirror that of human urinary and porcine-derived forms of KLK1. We believe that the results of this work suggest that the therapeutic action of DM199 will be the same or better than that of the forms marketed in Asia. In addition, there are also significant formulation, manufacturing, regulatory and other advantages for our synthetic human KLK1 drug candidate DM199:
|
● |
Potency and Impurity Considerations. KLK1 derived from human urine or porcine pancreas may contain impurities, endotoxins, and chemical byproducts due to the inherent variability of the isolation and purification process. We believe that this creates the risk of inconsistencies in potency and impurities from one production run to the next. However, we expect to produce a consistent formulation of KLK1 that is free of endotoxins and other impurities, which we believe will provide therapeutic benefits. |
|
● |
Cost and Scalability. Large quantities of human urine and porcine pancreas must be obtained to derive a small amount of KLK1. This creates potential procurement, cost and logistical challenges to source the necessary raw organic material, particularly for human urine sourced KLK1. Once sourced, the raw organic material is processed using chemicals and costly capital equipment and produces a significant amount of byproduct waste. Our novel recombinant manufacturing process utilizes widely available raw materials and can be readily scaled for commercial production. Accordingly, we believe our manufacturing process has significant cost and scalability advantages. |
|
● |
Regulatory. We are not aware of any attempts by manufacturers of the urine or porcine based KLK1 products to pursue regulatory approvals in the United States. We believe that this is related to challenges presented by using inconsistent and potentially hazardous biomaterials, such as human urine and porcine pancreas, and their resulting ability to produce a consistent drug product. Our novel recombinant manufacturing process utilizes widely available raw materials which we believe provides a significant regulatory advantage, particularly in regions such as the United States, Europe and Canada, where safety standards are high. In addition, we believe that DM199 could qualify for 12 years of data exclusivity under the Biologics Price Competition and Innovation Act of 2009, which was enacted as part of the ACA. |
Regulatory Approval
Securing regulatory approval for the manufacture and sale of human therapeutic products in the United States, Europe, Canada and other commercial territories is a long and costly process that is controlled by that particular territory’s national regulatory agency. The national regulatory agency in the United States is the FDA, in Europe it is EMA, and in Canada it is Health Canada. Other national regulatory agencies have similar regulatory approval processes, but each national regulatory agency has its own approval processes. Approval in the United States, Europe or Canada does not assure approval by other national regulatory agencies, although often test results from one country may be used in applications for regulatory approval in another country.
Prior to obtaining regulatory approval to market a drug product, every national regulatory agency has a variety of statutes and regulations which govern the principal development activities. These laws require controlled research and testing of products, governmental review, and approval of a submission containing preclinical and clinical data establishing the safety and efficacy of the product for each use sought, approval of manufacturing facilities including adherence to good manufacturing practices (“GMP”) during production and storage, and control of marketing activities, including advertising, labeling and pricing approval.
None of our product candidates have been completely developed or tested; and, therefore, we are not yet in a position to seek regulatory approval in any territory to market any of our product candidates.
The clinical testing, manufacturing, labeling, storage, distribution, record keeping, advertising, promotion, import, export, and marketing, among other things, of our product candidates are subject to extensive regulation by governmental authorities in the United States and other countries. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable requirements at any time during the product development process, approval process, or after approval may subject us to a variety of administrative or judicial sanctions, including refusal by the applicable regulatory authority to approve pending applications, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters and other types of letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement of profits, or civil or criminal investigations and penalties brought by the FDA and the Department of Justice or other governmental entities.
U.S. Approval Process
In the United States, the FDA, a federal government agency, is responsible for the drug approval process. The FDA’s mission is to ensure that all medications on the market are safe and effective. The FDA’s approval process examines potential drugs; and only those that meet strict requirements are approved.
The U.S. food and drug regulations require licensing of manufacturing facilities, carefully controlled research and testing of products, governmental review and approval of test results prior to marketing of therapeutic products, and adherence to GMP, as defined by each licensing jurisdiction, during production.
The drug approval process begins with the discovery of a potential drug. Pharmaceutical companies then test the drug extensively. A description of the different stages in the drug approval process in the United States follows.
Stage 1: Preclinical Research. After an experimental drug is discovered, research is conducted to help determine its potential for treating or curing an illness. This is called preclinical research. Animal and/or bench studies are conducted to determine if there are any harmful effects of the drug and to help understand how the drug works. Information from these experiments is submitted to the FDA in an IND. The FDA reviews the information in the IND and decides if the drug is safe to study in humans.
Stage 2: Clinical Research. In Stage 2, the experimental drug is studied in humans. The studies are known as clinical trials. Clinical trials are carefully designed and controlled experiments in which the experimental drug is administered to patients to test its safety and to determine the effectiveness of an experimental drug. The four general phases of clinical research are described below.
Phase I Clinical Studies. Phase I clinical studies are generally conducted with healthy volunteers who are not taking other medicines; patients with the illness that the drug is intended to treat are not tested at this stage. Ultimately, Phase I studies demonstrate how an experimental drug affects the body of a healthy individual. Phase I consists of a series of small studies consisting of “tens” of volunteers. Tests are done on each volunteer throughout the study to see how the person’s body processes, responds to, and is affected by the drug. Low doses and high doses of the drug are usually studied, resulting in the determination of the safe dosage range in volunteers by the end of Phase I. This information will determine whether the drug proceeds to Phase II.
Phase II Clinical Studies. Phase II clinical studies are conducted in order to determine how an experimental drug affects people who have the disease to be treated. Phase II usually consists of a limited number of studies that help determine the drug’s short-term safety, side effects, and general effectiveness. The studies in Phase II often are controlled investigations involving comparison between the experimental drug and a placebo, or between the experimental drug and an existing drug. Information gathered in Phase II studies will determine whether the drug proceeds to Phase III.
Phase III Clinical Studies. Phase III clinical studies are expanded controlled and uncontrolled trials that are used to more fully investigate the safety and effectiveness of the drug. These trials differ from Phase II trials because a larger number of patients are studied (sometimes in the thousands) and because the studies are usually of longer duration. As well, Phase Ill studies can include patients who have more than one illness and are taking medications in addition to the experimental drug used in the study. Therefore, the patients in Phase III studies more closely reflect the general population. The information from Phase III forms the basis for most of the drug’s initial labeling, which will guide physicians on how to use the drug.
Phase IV Clinical Studies. Phase IV clinical studies are conducted after a drug is approved. Companies often conduct Phase IV studies to more fully understand how their drug compares to other drugs. Also, the FDA may require additional studies after the drug is approved. FDA-required Phase IV studies often investigate the drug in specific types of patients that may not have been included in the Phase III studies and can involve very large numbers of patients to further assess the drug’s safety.
Stage 3: FDA Review for Approval. Following Phase III, the pharmaceutical company prepares reports of all studies conducted on the drug and a complete dossier on the manufacturing of the product and submits the reports to the FDA in a New Drug Application (“NDA”). The FDA reviews the information in the NDA to determine if the drug is safe and effective for its intended use. Occasionally, the FDA will ask experts for their opinion of the drug. If the FDA determines that the drug is safe and effective, the drug will be approved.
Stage 4: Marketing. After the FDA has approved the experimental drug, the pharmaceutical company can make it available to physicians and their patients. A company also may continue to conduct research to discover new uses for the drug. Each time a new use for a drug is discovered, the drug once again is subject to the entire FDA approval process before it can be marketed for that purpose.
Any pharmaceutical products for which FDA approvals are obtained are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with FDA promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, promoting pharmaceutical products for uses or in patient populations that are not described in the pharmaceutical product’s approved labeling (known as “off-label use”), industry-sponsored scientific and educational activities and promotional activities involving the internet. Failure to comply with FDA requirements can have negative consequences, including adverse publicity, enforcement letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal penalties.
The FDA also may require post-marketing testing, known as Phase IV testing, risk evaluation and mitigation strategies and surveillance to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product.
We believe that DM199 could qualify for 12 years of data exclusivity under the BPCIA, which was enacted as part of the ACA. Under the BPCIA, an application for a biosimilar product (“BLA”) cannot be submitted to the FDA until four years, or if approved by the FDA, until 12 years, after the original brand product identified as the reference product is approved under a BLA. The BPCIA provides an abbreviated pathway for the approval of biosimilar and interchangeable biological products. The new abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on its similarity to an existing brand product. The new law is complex and is only beginning to be interpreted and implemented by the FDA.
European Approval Process
The EMA is roughly parallel to the U.S. FDA in terms of the drug approval process and the strict requirements for approval. The EMA was set up in 1995 in an attempt to harmonize, but not replace, the work of existing national medicine regulatory bodies in individual European countries. As with the FDA, the EMA drug review and approval process follows different stages from preclinical testing through clinical testing in Phase I, II, and III. There are some differences between the FDA and EMA review process, specifically the review process in individual European countries. Such differences may allow certain drug products to be tested in patients at an earlier stage of development.
Other Healthcare Laws and Compliance Requirements
In the United States, our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including the Centers for Medicare and Medicaid Services and other divisions of the U.S. government, including, the Department of Health and Human Services, the U.S. Department of Justice and individual U.S. Attorney offices within the Department of Justice, and state and local governments. For example, if a drug product is reimbursed by Medicare, Medicaid, or other federal or state healthcare programs, our company, including our sales, marketing and scientific/educational grant programs, must comply with the federal False Claims Act, as amended, the federal Anti-Kickback Statute, as amended, and similar state laws. If a drug product is reimbursed by Medicare or Medicaid, pricing and rebate programs must comply with, as applicable, the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 (“OBRA”), and the Medicare Prescription Drug Improvement and Modernization Act of 2003. Among other things, OBRA requires drug manufacturers to pay rebates on prescription drugs to state Medicaid programs and empowers states to negotiate rebates on pharmaceutical prices, which may result in prices for our future products that will likely be lower than the prices we might otherwise obtain. Additionally, the ACA substantially changes the way healthcare is financed by both governmental and private insurers. There may continue to be additional proposals relating to the reform of the U.S. healthcare system, in the future, some of which could further limit coverage and reimbursement of drug products. If drug products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements may apply.
Pharmaceutical Coverage, Pricing and Reimbursement
In the United States and markets in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend in part on the availability of coverage and adequate reimbursement from third-party payers, including government health administrative authorities, managed care providers, private health insurers and other organizations. In the United States, private health insurers and other third-party payers often provide reimbursement for products and services based on the level at which the government (through the Medicare or Medicaid programs) provides reimbursement for such treatments. Third-party payers are increasingly examining the medical necessity and cost-effectiveness of medical products and services in addition to their safety and efficacy; and, accordingly, significant uncertainty exists as to the coverage and reimbursement status of newly approved therapeutics. In particular, in the United States, the European Union and other potentially significant markets for our product candidates, government authorities and third-party payers are increasingly attempting to limit or regulate the price of medical products and services, particularly for new and innovative products and therapies, which has resulted in lower average selling prices. Further, the increased emphasis on managed healthcare in the United States and on country and regional pricing and reimbursement controls in the European Union will put additional pressure on product pricing, reimbursement and usage, which may adversely affect our future product sales and results of operations. These pressures can arise from rules and practices of managed care groups, judicial decisions and governmental laws and regulations related to Medicare, Medicaid and healthcare reform, pharmaceutical reimbursement policies and pricing in general. As a result, coverage and adequate third party reimbursement may not be available for our products to enable us to realize an appropriate return on our investment in research and product development.
The market for our product candidates for which we may receive regulatory approval will depend significantly on access to third-party payers’ drug formularies or lists of medications for which third-party payers provide coverage and reimbursement. The industry competition to be included in such formularies often leads to downward pricing pressures on pharmaceutical companies. Also, third-party payers may refuse to include a particular branded drug in their formularies or may otherwise restrict patient access to a branded drug when a less costly generic equivalent or another alternative is available. In addition, because each third-party payer individually approves coverage and reimbursement levels, obtaining coverage and adequate reimbursement is a time-consuming and costly process. We would be required to provide scientific and clinical support for the use of any product candidate to each third-party payer separately with no assurance that approval would be obtained, and we may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost-effectiveness of our product candidates. This process could delay the market acceptance of any of our product candidates for which we may receive approval and could have a negative effect on our future revenues and operating results. We cannot be certain that our product candidates will be considered cost-effective. If we are unable to obtain coverage and adequate payment levels for our product candidates from third-party payers, physicians may limit how much or under what circumstances they will prescribe or administer them and patients may decline to purchase them. This in turn could affect our ability to successfully commercialize our products and impact our profitability, results of operations, financial condition, and future success.
Research and Development
We have devoted substantially all of our efforts to research and development (“R&D”) which therefore comprises the largest component of our operating costs. Our primary focus over the past approximately eight years has been our lead product candidate, DM199, which is currently in clinical development for AIS and is expected to commence clinical development for CKD in late 2018 or first half of 2019.
We expect our R&D expenses will continue to increase in the future as we advance our initial product candidate through clinical trials in AIS and CKD and seek to expand our product candidate portfolio. The process of conducting the necessary clinical research to obtain regulatory approval is costly and time-consuming and we consider the active management and development of our clinical pipeline to be integral to our long-term success. The actual probability of success for each product candidate, clinical indication and preclinical program may be affected by a variety of factors including, among other things, the safety and efficacy data for product candidates, amounts invested in the program, competition and competitive developments, manufacturing capability and commercial viability.
Research and development expenses include:
|
● |
expenses incurred under contract research agreements and other agreements with third parties; |
|
● |
expenses incurred under agreements with clinical trial sites that conduct research and development activities on our behalf; |
|
● |
employee and consultant-related expenses, which include salaries, benefits, travel and share-based compensation; |
|
● |
laboratory and vendor expenses related to the execution of clinical trials and non-clinical studies; |
|
● |
the cost of acquiring, developing, manufacturing, and distributing clinical trial materials; and |
|
● |
facilities and other expenses, which include direct and allocated expenses for rent and maintenance of facilities, insurance, and other supply costs. |
Research and development costs are expensed as incurred. Costs for certain development activities such as clinical trials are recognized based on an evaluation of the progress to completion of specific tasks using information and data provided to us by our vendors and our clinical sites.
We expect that it will be several years, if ever, before we have any product candidates ready for commercialization. Our research and development expenses totaled $3.2 million and $1.7 million in 2017 and 2016, respectively.
Manufacturing
We do not own or operate manufacturing facilities for the production of clinical or commercial quantities of DM199 nor do we have plans to develop our own manufacturing operations in the foreseeable future. We rely on Catalent for all of our required raw materials, active pharmaceutical ingredients and finished DM199 product candidate for our clinical trials. We have licensed certain gene expression technology and we contract with Catalent for the manufacture of DM199. The royalty term is indefinite but may be canceled by us on 90 days’ prior written notice. The license may not be terminated by Catalent unless we fail to make required milestone and royalty payments. We currently employ internal resources and third-party consultants to manage our manufacturing relationship with Catalent.
Sales and Marketing
We have not yet defined our sales, marketing or product distribution strategy for our initial product candidate, or any future product candidates, because it is still early in the clinical development stage. We currently expect to partner with a large pharmaceutical company for sales execution. However, our future commercial strategy may include the use of distributors, a contract sales force or the establishment of our own commercial and specialty sales force, as well as similar strategies for regions and territories outside the United States.
Intellectual Property
We view patents and other means of intellectual property protection including trade secrets as an important component of our core business. We focus on translating our innovations into intangible property protecting our proprietary technology from infringement by competitors. To that end, patents are reviewed frequently and continue to be sought in relation to those components or concepts of our preclinical and clinical products to provide protection. Our strategy, where possible, is to file patent applications to protect our product candidates, as well as methods of manufacturing, administering and using a product candidate. Prior art searches of both patent and scientific databases are performed to evaluate novelty, inventiveness and freedom-to-operate. We require all employees, consultants, and parties to sign a collaborative research agreement and to execute confidentiality agreements upon the commencement of employment, consulting relationships, or a collaboration with us. These agreements require that all confidential information developed or made known during the course of the engagement with us is to be kept confidential. We also maintain agreements with our scientific staff and all parties contracted in a scientific capacity affirming that all inventions resulting from work performed for us, using our property, or relating to our business and conceived or completed during the period covered by the agreement are the exclusive property of our company.
Our patent portfolio includes patents and pending applications that are owned by us, which include claims for composition of matter and methods of use. For our DM199 program, this includes two patent families that are directed to composition of matter, and methods of use.
The DM199 patents protect composition of matter including compositions of glycoforms, formulations, methods of administration and a variety of therapeutic approaches pertaining to current and potential future indications. We currently have additional patent applications for DM199. Additionally, for the manufacture of DM199, we have licensed an expression system and cell line with proven GMP and regulatory support and are contracting with a contract manufacturing organization (“CMO”) with proven GMP experience in manufacturing of recombinant proteins for clinical trials.
We do not own or operate manufacturing facilities for the production of clinical or commercial quantities of DM199. We intend to rely on Catalent for the manufacture of DM199. We have licensed certain gene expression technology and we contract with Catalent for the manufacture of DM199. Under the terms of this license, certain milestone and royalty payments may become due and are dependent upon, among other factors, performing clinical trials, obtaining regulatory approvals and ultimately the successful development of a new drug, the outcome and timing of which is uncertain. The royalty term is indefinite but may be canceled by us on 90 days’ prior written notice. The license may not be terminated by Catalent unless we fail to make required milestone and royalty payments.
Our DM199 patent portfolio includes granted U.S. patents, a granted European patent, one pending U.S. patent application and a worldwide pending application filed under the Patent Cooperation Treaty (“PCT”). Granted or pending claims offer various forms of protection for DM199 including claims to compositions of matter, pharmaceutical compositions, specific formulations and dosing levels, and methods for treating a variety of diseases, including stroke, chronic kidney disease, and related disorders. These U.S. patents and applications, and their foreign equivalents, are described in more detail below.
Issued patents held by us cover the DM199 composition of matter based on an optimized combination of closely-related isoforms that differ in the extent of glycosylation (process by which sugars are chemically attached to proteins). Issued claims in this patent family cover the most pharmacologically active variants of DM199 and methods of using the same for treating ischemic conditions and these patents are due to expire in 2033. A second patent family includes an issued U.S. patent with claims directed to methods of treating subjects by administering a subcutaneous formulation of DM199 or related recombinant kallikrein-1 polypeptides. The PCT patent application is directed to a range of dose levels and dosing regimens of DM199 that are potentially useful for treating a wide range of diseases including, e.g. pulmonary arterial hypertension, cardiac ischemia, chronic kidney disease, diabetes, stroke, and vascular dementia.
Methods and reagents required for commercial scale manufacture of DM199 are subject to a series of patents issued to our manufacturing partner. As noted above, we exclusively license these patents from our manufacturing partner for the production of DM199 or any human KLK1 protein. We believe that our proprietary technology along with trade secrets will provide substantial protection from third-party competitors. We believe DM199 cannot be reversed engineered for a copycat version to be made. In addition, DiaMedica has specialized knowledge of the manufacturing process.
We believe that the most relevant granted patents with composition of matter or method of use claims covering DM199 are listed below, along with their projected expiration dates exclusive of any patent term extension.
|
Patent Number |
Title |
Geography |
Expiration |
|||
|
Issued patents |
|
|
|
|||
|
US 9,364,521 |
Composition of Matter – Human Tissue Kallikrein 1 Glycosylation Isoforms |
US |
2033 |
|||
|
EP 2 854 841 |
Composition of Matter – Human Tissue Kallikrein 1 Glycosylation Isoforms |
Europe |
2033 |
|||
|
US 9,616,015 |
Formulations for Human Tissue Kallikrein-1 for Parenteral Delivery and Related Methods |
US |
2033 |
|||
|
Pending applications |
|
|
|
|||
|
PCT/US2018/021749 |
Dosage Forms of Tissue Kallikrein 1 |
US/Worldwide |
2038 |
License Agreement
In September 2018, we entered into a license and collaboration agreement with Ahon Pharma, which grants Ahon Pharma exclusive rights to develop and commercialize DM199 for acute ischemic stroke in mainland China, Taiwan, Hong Kong S.A.R. and Macau S.A.R. Under the terms of the agreement, we are entitled to receive an upfront payment of $5.0 million, consisting of $500,000, on signing and $4.5 million upon regulatory clearance to initiate a clinical trial in China. We also have the potential to receive up to an additional $27.5 million in development and sales related milestones and up to approximately 10% royalties on net sales of DM199 in the licensed territories. All development, regulatory, sales, marketing, and commercial activities and associated costs in the licensed territories will be the sole responsibility of Ahon Pharma. This agreement may be terminated at any time by Ahon Pharma by providing 120 days written notice. Fosun Pharma, through its partnership with SK Group, a South Korea based company is an investor in DiaMedica through its equity investment in 2016.
Legal Proceedings
In March 2013, we entered into a clinical research agreement with PRA to perform a double-blinded, placebo-controlled, single-dose and multiple-dose study to evaluate the safety, tolerability, pharmacokinetics, pharmacodynamics and proof of concept of DM199 in healthy subjects and in patients with Type 2 diabetes mellitus. In one arm of this study, we enrolled 36 patients with Type 2 diabetes who were treated with two subcutaneous dose levels of DM199 over a 28-day period. This study achieved its primary endpoint and demonstrated that DM199 was well-tolerated. The secondary endpoints for this study, however, were not met. The secondary efficacy endpoints were confounded due to what we believe were significant execution errors caused by protocol deviations occurring at the clinical trial site that were unable to be reconciled. To date, we have been unable to obtain the complete study records from PRA and generate a final study report. On November 14, 2017, we initiated litigation with PRA in the United States District Court, Southern District of New York, to compel them to comply with the terms of the clinical research agreement, including providing full study records and to recover damages. After PRA objected to the venue, on August 24, 2018, we re-filed our complaint against PRA in the United States District Court, District of Delaware. The complaint alleges, among other things, that PRA failed to conduct the study in accordance with the study protocol and with generally accepted standards for conducting such clinical trials and that PRA further refused to provide us with all data, records and documentation, and/or access thereto, related to the study in accordance with the clinical trial study agreement. The complaint seeks to compel PRA to comply with the terms of the clinical trial study agreement, including providing full study records and to recover damages.
From time to time, we may be subject to other various ongoing or threatened legal actions and proceedings, including those that arise in the ordinary course of business, which may include employment matters and breach of contract disputes. Such matters are subject to many uncertainties and to outcomes that are not predictable with assurance and that may not be known for extended periods of time. In the opinion of management, the outcome of such routine ongoing litigation is not expected to have a material adverse effect on our results of operations or financial condition.
Facilities
Our principal executive offices, together with our research and development operations, are at the office of our wholly owned subsidiary, DiaMedica USA Inc., located at 2 Carlson Parkway, Suite 260, Minneapolis, Minnesota, USA 55447. We lease these premises, which consist of approximately 3,800 square feet, pursuant to a lease that expires in August 2022. We believe that our facilities are adequate for our current needs and that suitable additional space will be available as and when needed on acceptable terms.
Employees
As of November 15, 2018, we had 11 full-time employees. We have never had a work stoppage and none of our employees are covered by collective bargaining agreements. We believe our employee relations are good.
Enforceability of Civil Liabilities Against Foreign Persons
We are organized under and governed by the federal laws of Canada, and, accordingly, are governed by the applicable laws of Canada. There is doubt as to the enforceability, in original actions in Canadian courts, of liabilities based upon the U.S. federal securities laws or the securities laws or “blue sky” laws of any state within the United States and as to the enforceability in Canadian courts of judgments of U.S. courts obtained in actions based upon the civil liability provisions of the U.S. federal securities laws or any such state securities laws or blue sky laws. Accordingly, it may not be possible to enforce judgments obtained in the United States against us.
MANAGEMENT
Executive Officers and Directors
The following table sets forth information as of November 15, 2018 regarding each of our current executive officers and directors:
|
Name |
Age |
Positions |
||
|
Rick Pauls |
47 |
President and Chief Executive Officer, Director |
||
|
Scott Kellen |
53 |
Chief Financial Officer and Secretary |
||
|
Todd Verdoorn, Ph.D. |
57 |
Chief Scientific Officer |
||
|
Harry Alcorn, Pharm.D. |
62 |
Chief Medical Officer |
||
|
Richard Pilnik(1)(2)(3) |
61 |
Chairman of the Board |
||
|
Michael Giuffre, M.D.(1)(2)(3) |
63 |
Director |
||
|
James Parsons(1)(2)(3) |
53 |
Director |
||
|
Zhenyu Xiao, Ph.D. |
44 |
Director |
______________________________
|
(1) |
Member of the Audit Committee. |
|
(2) |
Member of the Compensation Committee. |
|
(3) |
Member of the Governance and Nominating Committee. |
The present principal occupations and recent employment history of each of our executive officers and directors are set forth below. Pursuant to the CBCA, at least 25% of our directors must be resident Canadians.
Executive Officers
Rick Pauls was appointed our President and Chief Executive Officer in January 2010. Mr. Pauls has served as a member of our Board of Directors since April 2005 and the Chairman of the Board from April 2008 to July 2014. Prior to joining DiaMedica, Mr. Pauls was the Co-Founder and Managing Director of CentreStone Ventures Inc., a life sciences venture capital fund, from February 2002 until January 2010. Mr. Pauls was an analyst for Centara Corporation, another early stage venture capital fund, from January 2000 until January 2002. From June 1997 until November 1999, Mr. Pauls worked for General Motors Acceptation Corporation specializing in asset-backed securitization and structured finance. Mr. Pauls previously served as an independent member of the board of directors of LED Medical Diagnostics, Inc. Mr. Pauls received his Bachelor of Arts in Economics from the University of Manitoba and his M.B.A. in Finance from the University of North Dakota.
We believe that Mr. Pauls’s experience in the biopharmaceutical industry as an executive and investor and his extensive knowledge of all aspects of our company, business, industry, and day-to-day operations as a result of his role as our President and Chief Executive Officer enable him to make valuable contributions to our Board of Directors. In addition, as a result of his role as President and Chief Executive Officer, Mr. Pauls provides unique insight into our future strategies, opportunities and challenges, and serves as the unifying element between the leadership and strategic direction provided by our Board of Directors and the implementation of our business strategies by management.
Scott Kellen was appointed our Chief Financial Officer and Secretary in April 2018. Prior to joining DiaMedica, Mr. Kellen served as Vice President and Chief Financial Officer of Sun BioPharma, Inc., a publicly-traded clinical stage drug development company, from October 2015 until April 2018. From February 2010 to September 2015, Mr. Kellen served as Chief Financial Officer and Secretary of Kips Bay Medical, Inc., a publicly-traded medical device company, and became Chief Operating Officer of Kips Bay in March 2012. From November 2007 to May 2009, Mr. Kellen served as Finance Director of Transoma Medical, Inc. From 2005 to October 2007, Mr. Kellen served as Corporate Controller of ev3 Inc. From March 2003 to April 2005, Mr. Kellen served as Senior Manager, Audit and Advisory Services of Deloitte & Touche, LLP. Altogether, Mr. Kellen has spent more than 25 years in the life sciences industry, focusing on publicly traded early stage and growth companies. Mr. Kellen has a Bachelor of Science degree in Business Administration from the University of South Dakota and is a Certified Public Accountant (inactive).
Todd Verdoorn, Ph.D. was appointed our Chief Scientific Officer in May 2016. From January 2016 to April 2016, Dr. Verdoorn served as our Vice President, Neuroscience. Prior to joining DiaMedica, Dr. Verdoorn served as Chief Scientist at Intuitive Quantitation, LLC, a company that provides strategic and tactical leadership for companies creating new treatments, from May 2013 to December 2016. From September 2011 to May 2013, Dr. Verdoorn served as Vice President, Neurobiology at NeuroTherapeutics Pharma, Inc., a company that develops and markets therapeutics. From January 2008 to August 2011, Dr. Verdoorn served as Chief Scientist for Orasi Medical, Inc., a medical device company. From June 2007 to January 2008, Dr. Verdoorn served as Chief Scientific Officer for Smart Bioscience SAS, a company that discovers and develops small-molecule therapeutics. Prior to joining Smart Bioscience, Dr. Verdoorn served as Chief Scientific Officer at Algos Preclinical Services, Inc., a research and consulting company, from January 2003 to June 2007. Dr. Verdoorn has more than 26 years of experience working with both public and private companies to develop new treatments for neurological diseases, including five years working with Bristol-Myers Squibb’s stroke group. Dr. Verdoorn has a Bachelor of Arts degree in Chemistry from Central College and he earned his Ph.D. in Neurobiology from the University of North Carolina, conducting his post-doctoral research at the Max Planck Institute with Nobel Laureate Dr. Bert Sakmann and served as Associate Professor of Pharmacology at Vanderbilt University School of Medicine.
Harry Alcorn Jr. Pharm.D. was appointed our Chief Medical Officer in August 2018. Prior to joining DiaMedica, Dr. Alcorn served as Chief Scientific Officer at DaVita Clinical Research (“DCR”), a company that provides clinical research services for Pharmaceutical and Biotech companies, from October 1997 to June 2018. While at DCR, Dr. Alcorn was responsible for clinical research operations, including the formation and management of the early clinical and late phase research services. Dr. Alcorn also founded the U.S. Renal Network, the first network of Phase I renal research sites in the United States. Dr. Alcorn developed DCR’s site management organization for clinical trials. Dr. Alcorn also served as an Executive Director, a Pharmacist and an Investigator at DCR. During this time, from Jan 2013 to December 2014, he also served on the Board of Directors for the Association of Clinical Pharmacology Units, an association of Phase I clinical trial sites. Dr. Alcorn has over 30 years of clinical research experience working with Biotech and Pharmaceutical companies, both public and private, in conducting research in renal, hepatic and cardiovascular disease. Dr. Alcorn has written and consulted on the development of several protocols and has served as Principal Investigator or Sub Investigator in numerous studies and, for several of these studies, presented study design and results to the FDA. Currently he holds clinical faculty appointments with the University of Minnesota, Creighton University, University of Nebraska Medical Center, Virginia Commonwealth and the University of Colorado, Denver. Dr. Alcorn graduated from Creighton University with a Bachelor of Pharmacy and went on to earn his Doctor of Pharmacy degree from University of Nebraska Medical Center.
Non-Employee Directors
Richard Pilnik has served as a member of our board of directors since May 2009. Mr. Pilnik serves as our Chairman of the Board. Mr. Pilnik has served as the President and member of the board of directors of Vigor Medical Services, Inc., a medical device company, since May 2017. From December 2015 to November 2017, Mr. Pilnik served as a member of the board of directors of Chiltern International Limited, a private leading mid-tier Clinical Research Organization, and was Chairman of the Board from April 2016 to November 2017. Mr. Pilnik has a 30-year career in healthcare at Eli Lilly and Company, a pharmaceutical company, and Quintiles Transnational Corp., a global pioneer in pharmaceutical services. From April 2009 to June 2014, Mr. Pilnik served as Executive Vice President and President of Quintiles Commercial Solutions, an outsourcing business to over 70 pharma and biotech companies. Prior to that, he spent 25 years at Eli Lilly and Company where he held several leadership positions, most recently as Group Vice President and Chief Marketing Officer from May 2006 to July 2008. Mr. Pilnik was directly responsible for commercial strategy, market research, new product planning and the medical marketing interaction. From December 2000 to May 2006, Mr. Pilnik served as President of Eli Lilly Europe, Middle East and Africa and the Commonwealth of Independent States, a regional organization of former Soviet Republics, and oversaw 50 countries and positioned Eli Lilly as the fastest growing pharmaceutical company in the region. Mr. Pilnik also held several marketing and sales management positions in the United States, Europe and Latin America. Mr. Pilnik currently serves on the board of directors of Vigor Medical Systems, Inc., NuSirt, an early-stage biopharma, and the Duke University Fuqua School of Business. Mr. Pilnik previously served on the board of directors of Elan Pharmaceuticals, Chiltern International, the largest mid-size Clinical Research Organization, and Certara, L.P., a private biotech company focused on drug development modeling and biosimulation. Mr. Pilnik holds a Bachelor of Arts in Economics from Duke University and an MBA from the Kellogg School of Management at Northwestern University.
We believe that Mr. Pilnik’s deep experience in the industry and his history and knowledge of our company enable him to make valuable contributions to our Board of Directors.
Michael Giuffre, M.D. has served as a member of our Board of Directors since August 2010. Since July 2009, Dr. Giuffre has served as a Clinical Professor of Cardiac Sciences and Pediatrics at the University of Calgary and has had an extensive portfolio of clinical practice, cardiovascular research and university teaching. Dr. Giuffre is actively involved in health care delivery, medical leadership and in the biotechnology business sector. Since 2012, Dr. Giuffre has served as the Chief Scientific Officer and a member of the board of directors of FoodChek Systems Inc. and in November 2017, he became Chairman of the Board. Dr. Giuffre also serves as President of FoodChek Laboratories Inc. Dr. Giuffre previously served on the board of directors of the Canadian Medical Association (CMA), Unicef Canada, the Alberta Medical Association (AMA), Can-Cal Resources Ltd, Vacci-Test Corporation, IC2E International Inc. and MedMira Inc. Dr. Giuffre has received a Certified and Registered Appointment and a Distinguished Fellow appointment by the American Academy of Cardiology (FACC). In 2005, he was awarded Physician of the Year by the Calgary Medical Society and in 2017 was “Mentor of the Year” for the Royal College of Physicians and Surgeons of Canada. Dr. Giuffre was also a former President of the AMA and the Calgary and Area Physicians Association and also a past representative to the board of the Calgary Health Region. Dr. Giuffre holds a Bachelor of Science in cellular and microbial biology, a Ph.D. candidacy in molecular virology, an M.D. and an M.B.A. He is Canadian Royal College board certified in specialties that include Pediatrics and Pediatric Cardiology and has a subspecialty in Pediatric Cardiac Electrophysiology. Dr. Giuffre is a member of the board of directors of Avenue Living, a private real estate company in Calgary, Alberta, Canada and its affiliates, Avondale Real Estate Capital Ltd. and AgriSelect Land Capital, Ltd., both private real estate companies in Calgary, Alberta Canada. Dr. Giuffre is a resident of Canada.
We believe that Dr. Giuffre’s medical experience, including as a practicing physician and professor, enable him to make valuable contributions to our Board of Directors.
James Parsons has served as a member of our Board of Directors since October 2015. Previously, Mr. Parsons served as our Vice President of Finance from October 2010 until May 2014. Since August 2011, Mr. Parsons has served as Chief Financial Officer and Corporate Secretary of Trillium Therapeutics Inc., a Nasdaq-listed immuno-oncology company. Mr. Parsons serves as a member of the board of directors and audit committee chair of Sernova Corp., which is listed on the TSX Venture Exchange. Mr. Parsons has been a Chief Financial Officer in the life sciences industry since 2000 with experience in therapeutics, diagnostics and devices. Mr. Parsons has a Master of Accounting degree from the University of Waterloo and is a Chartered Professional Accountant and Chartered Accountant. Mr. Parsons is a resident of Canada.
We believe that Mr. Parsons’ financial experience, including his history and knowledge of our company, enable him to make valuable contributions to our Board of Directors.
Zhenyu Xiao, Ph.D. has served as a member of our Board of Directors since November 2016. Dr. Xiao was elected to our Board of Directors in connection with the equity investment by Hermeda Industrial Co., Limited and is its designee to the Board of Directors under an investment agreement which is described in more detail under “Item 7. Certain Relationships and Related Party Transactions.” Dr. Xiao has been the Chief Executive Officer of Hermed Equity Investment Management (Shanghai) Co., Ltd., a private equity fund. From June 2008 to November 2014, Dr. Xiao was the Associate General Manager of Shanghai Fosun Pharmaceutical Group Co Ltd., a pharmaceutical manufacturing company, where he was the deputy chief of the IPO team for the Fosun Pharma Listing in Hong Kong Exchange and the deputy director of Fosun Pharmaceutical Technological Center in charge of evaluating new technology and R&D and investment. Dr. Xiao has a Ph.D. degree in Pharmacology and conducted his postdoctoral research at University of Rochester (NY), co-founding a pharmaceutical company with Dr. Paul Okunieff and winning Small Business Technology Transfer support, a U.S. Small Business Administration program to facilitate joint venture opportunities between small businesses and non-profit research institutions.
We believe that Dr. Xiao’s experience in the industry, including as an investor, enable him to make valuable contributions to our Board of Directors.
Family Relationships
No family relationships exist among any of our directors or executive officers.
Involvement in Certain Legal Proceedings
To the best of our knowledge, none of our directors or executive officers have, during the past ten years, been involved in any legal proceedings described in subparagraph (f) of Item 401 of Regulation S-K.
Board Composition
Our Board of Directors consists of five directors, four of whom qualify as independent directors under the rules and regulations of the SEC and The Nasdaq Capital Market. Pursuant to the CBCA, at least 25% of our directors must be resident Canadians.
Election of Directors
Our bylaws provide that members of our Board or Directors are elected by a majority of votes cast by our shareholders.
Independence of our Board and Board Committees
Rule 5605 of the Nasdaq Marketplace Rules (“Nasdaq Listing Rules”) requires a majority of a listed company’s board of directors be “independent” as defined in Nasdaq Listing Rule 5605(a)(2) within one year of listing. In addition, the Nasdaq Listing Rules require that, subject to specified exceptions, each member of a listed company’s audit, compensation, and nominating and corporate governance committees be independent and that audit committee members also satisfy independence criteria set forth in Rule 10A-3 under the Exchange Act.
Based upon information requested from and provided by each director concerning his background, employment and affiliations, including family and other relationships, including those relationships described under “Certain Relationships and Related Party Transactions,” we believe that none of our non-employee directors, representing four of our five directors, have a relationship that interferes with the exercise of independent judgment in carrying out the responsibilities of a director and that each of these directors is “independent” as that term is defined under Rule 5605(a)(2) of the Nasdaq Listing Rules. Rick Pauls is not considered independent because of his service as our Chief Executive Officer.
Each director who serves as a member of the audit, compensation, and nominating and corporate governance committees satisfies the independence standards for such committees established by the SEC and the Nasdaq Listing Rules, as applicable. In making these determinations on the independence of our directors, our Board of Directors has considered the relationships that each such non-employee director has with the company and all other facts and circumstances our Board of Directors deemed relevant in determining independence, including the beneficial ownership of our common shares by each non-employee director.
Leadership Structure of the Board
Under the corporate governance guidelines, the Board of Directors may select from its members a Chairman of the Board. The office of Chairman of the Board and the office of President and Chief Executive may or may not be held by one person. The Board believes it is best not to have a fixed policy on this issue and that it should be free to make this determination based on what it believes is best in the circumstances. The Nominating and Corporate Governance Committee will review periodically the leadership structure of the Board of Directors to assess whether it is appropriate given the specific characteristics and circumstances of the company. However, the Board of Directors does strongly endorse the concept of independent directors being in a position of leadership for the rest of the independent directors. If at any time, the Chief Executive Officer and Chairman of the Board are the same, the Board of Directors shall elect an independent director to serve as the lead director. The lead director will have the following duties and responsibilities in addition to such other duties and responsibilities as may be determined by the Board of Directors from time to time:
|
● |
chairing the executive sessions of the independent directors and calling meetings of the independent directors; |
|
● |
determining the agenda for the executive sessions of the independent directors, and participating with the Chairman of the Board in establishing the agenda for Board meetings; |
|
● |
coordinating feedback among the independent directors and the Chief Executive Officer; |
|
● |
overseeing the development of appropriate responses to communications from shareholders and other interested persons addressed to the independent directors as a group; |
|
● |
on behalf of the independent directors, retaining legal counsel or other advisors as they deem appropriate in the conduct of their duties and responsibilities; and |
|
● |
performing such other duties as the Board of Directors deems appropriate from time to time. |
Mr. Pilnik currently serves as Chairman of the Board and Rick Pauls currently serves as President and Chief Executive Officer.
We currently believe this leadership structure is in the best interests of DiaMedica and our shareholders and strikes the appropriate balance between the President and Chief Executive Officer’s responsibility for the strategic direction, day-to day-leadership and performance of our company and the Chairman of the Board’s responsibility to guide overall strategic direction of our company and provide oversight of our corporate governance and guidance to our President and Chief Executive Officer and to set the agenda for and preside over board meetings. We recognize that different leadership structures may be appropriate for companies in different situations and believe that no one structure is suitable for all companies. We believe that our company is well-served by this leadership structure. We anticipate that our Board of Directors will periodically review our leadership structure and may make such changes in the future as it deems appropriate.
Board Committees
Our Board of Directors has three standing committees: an Audit Committee, a Compensation Committee and a Nominating and Corporate Governance Committee. Each of these committees has the composition described in the table below and the responsibilities described in the sections below. Our Board of Directors has adopted a written charter for each committee of our Board of Directors. Our Board of Directors from time to time may establish other committees.
The following table summarizes the current membership of each of our three board committees.
|
Director |
Audit Committee |
Compensation Committee |
Nominating and Corporate Governance Committee |
|||
|
Rick Pauls |
— |
— |
— |
|||
|
Michael Giuffre, M.D. |
√ |
Chair |
√ |
|||
|
James Parsons |
Chair |
√ |
√ |
|||
|
Richard Pilnik |
√ |
√ |
Chair |
|||
|
Zhenyu Xiao, Ph.D. |
— |
— |
— |
Audit Committee
The Audit Committee assists the Board of Directors in fulfilling its oversight responsibilities relating to our annual and quarterly financial statements filed with the SEC and any applicable securities regulatory authorities of the provinces and territories of Canada, our financial reporting process, our internal control over financial accounting and disclosure controls and procedures, the annual independent audit of our financial statements, and the effectiveness of our legal compliance and ethics programs. The Audit Committee’s primary responsibilities include:
|
● |
overseeing our financial reporting process, internal control over financial reporting and disclosure controls and procedures on behalf of the Board of Directors; |
|
● |
having sole authority to appoint, oversee, evaluate, retain and terminate the engagement of our independent registered public accounting firm and establish the compensation to be paid to the firm; |
|
● |
reviewing and pre-approving all audit services and permissible non-audit services to be provided to us by our independent registered public accounting firm; |
|
● |
establishing procedures for the receipt, retention and treatment of complaints regarding accounting, internal accounting controls or auditing matters and for the confidential, anonymous submission by our employees of concerns regarding questionable accounting or auditing matters; and |
|
● |
overseeing our systems to monitor legal and ethical compliance programs, including the establishment and administration of (including the grant of any waiver from) a written code of ethics applicable to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. |
The Audit Committee has the authority to engage the services of outside experts and advisors as it deems necessary or appropriate to carry out its duties and responsibilities.
Each member of the Audit Committee qualifies as “independent” for purposes of membership on audit committees pursuant to the Nasdaq Listing Rules and the rules and regulations of the SEC and is “financially literate” as required by the Nasdaq Listing Rules. In addition, the Board of Directors has determined that Mr. Parsons qualifies as an “audit committee financial expert” as defined by the rules and regulations of the SEC and meets the qualifications of “financial sophistication” under the Nasdaq Listing Rules as a result of his extensive financial background and various financial positions he has held throughout his career. Shareholders should understand that these designations related to Audit Committee members’ experience and understanding with respect to certain accounting and auditing matters do not impose upon any of them any duties, obligations or liabilities that are greater than those generally imposed on a member of the Audit Committee or of the Board of Directors.
Compensation Committee
The Compensation Committee assists the Board of Directors in fulfilling its oversight responsibilities relating to the compensation of our Chief Executive Officer and other executive officers and administers our equity compensation plans. The Compensation Committee’s primary responsibilities include:
|
● |
determining all compensation for our Chief Executive Officer and other executive officers; |
|
● |
reviewing, assessing and approving overall strategies for attracting, developing, retaining and motivating our management and employees; |
|
● |
overseeing the development and implementation of succession plans for our Chief Executive Officer and other key executive officers and employees; |
|
● |
reviewing, assessing and approving overall compensation structure on an annual basis; and |
|
● |
recommending and leading a process for the determination of non-employee director compensation. |
The Compensation Committee has the authority to engage the services of outside experts and advisors as it deems necessary or appropriate to carry out its duties and responsibilities, and prior to doing so, assesses the independence of such experts and advisors from management.
The Board of Directors has determined that each of the members of the Compensation Committee is considered an “independent director” under the Nasdaq Listing Rules and a “non-employee director” within the meaning of Rule 16b-3 under the Exchange Act.
Nominating and Corporate Governance Committee
The Nominating and Corporate Governance Committee assists the Board of Directors in fulfilling its oversight responsibilities relating to director nominations and corporate governance. The primary responsibilities of the Nominating and Corporate Governance Committee include:
|
● |
identifying individuals qualified to become members of the Board of Directors, which includes reviewing and considering director nominees submitted by shareholders; |
|
● |
recommending director nominees for each annual general meeting of shareholders and director nominees to fill any vacancies that may occur between general meetings of shareholders; |
|
● |
being aware of best practices in corporate governance matters and developing and recommending to the Board of Directors a set of corporate governance guidelines to govern the Board of Directors, its committees, the company and our employees; and |
|
● |
developing and overseeing an annual Board of Directors and Board committee evaluation process. |
The Nominating and Corporate Governance Committee has the authority to engage the services of outside experts and advisors as it deems necessary or appropriate to carry out its duties and responsibilities.
The Board of Directors has determined that each of the members of the Nominating and Corporate Governance Committee is considered an “independent director” under the Nasdaq Listing Rules.
Board Diversity
The Nominating and Corporate Governance Committee is responsible for reviewing with our Board of Directors, on an annual basis, the appropriate characteristics, skills and experience required for our Board of Directors as a whole and its individual members. In evaluating the suitability of individual candidates (both new candidates and current members), the Nominating and Corporate Governance Committee, in recommending candidates for election, and our Board of Directors, in approving (and, in the case of vacancies, appointing) such candidates, take into account many factors, including the following:
|
● |
personal and professional integrity, ethics and values; |
|
● |
experience in corporate management, such as serving as an officer or former officer of a publicly held company; |
|
● |
strong finance experience; |
|
● |
relevant social policy concerns; |
|
● |
experience relevant to our industry; |
|
● |
experience as a board member or executive officer of another publicly held company; |
|
● |
relevant academic expertise or other proficiency in an area of our operations; |
|
● |
diversity of expertise and experience in substantive matters pertaining to our business relative to other board members; |
|
● |
diversity of background and perspective, including, but not limited to, with respect to age, gender, race, place of residence and specialized experience; |
|
● |
practical and mature business judgment, including, but not limited to, the ability to make independent analytical inquiries; |
|
● |
geographic location, in light of the fact that at least 25% of our directors must be Canadian residents; and |
|
● |
any other relevant qualifications, attributes or skills. |
The Board of Directors evaluates each individual in the context of the Board of Directors as a whole, with the objective of assembling a group that can best perpetuate the success of the business and represent shareholder interests through the exercise of sound judgment using its diversity of experience in these various areas. In determining whether to recommend a director for re-election, the Nominating and Corporate Governance Committee may also consider the director’s past attendance at meetings and participation in and contributions to the activities of the Board of Directors.
Code of Business Conduct and Ethics
We have adopted a code of business conduct and ethics applicable to all of our directors, officer and employees, in accordance with Section 406 of the Sarbanes-Oxley Act, the rules of the SEC promulgated thereunder, and the Nasdaq Listing Rules. In the event that any changes are made or any waivers from the provisions of the code of business conduct and ethics are made, these events would be disclosed on our website or in a report on Form 8-K within four business days of such event. The code of business conduct and ethics is posted on our website at www.diamedica.com. Copies of the code of business conduct and ethics will be provided free of charge upon written request directed to Investor Relations, DiaMedica Therapeutics Inc. 2 Carlson Parkway, Suite 260, Minneapolis, Minnesota 55447.
Role of Board in Risk Oversight Process
Risk is inherent with every business. We face a number of risks, including regulatory, compliance, legal, competitive, financial (accounting, credit, interest rate, liquidity and tax), operational, political, strategic and reputational risks. Our management is responsible for the day-to-day management of risks faced by us, while our Board of Directors, as a whole and through its committees, has responsibility for the oversight of risk management. In its risk oversight role, our Board of Directors ensures that the risk management processes designed and implemented by management are adequate and functioning as designed. Our Board of Directors oversees risks through the establishment of policies and procedures that are designed to guide daily operations in a manner consistent with applicable laws, regulations and risks acceptable to us. Our President and Chief Executive Officer, who is also a board member, regularly discusses with the Board of Directors the strategies and risks facing our company.
The standing committees of the Board of Directors oversee risks associated with their respective principal areas of focus. The Audit Committee’s role includes a particular focus on the qualitative aspects of financial reporting to shareholders, on our processes for the management of business and financial risk. The Audit Committee, along with management, is also responsible for developing and participating in a process for review of important financial and operating topics that present potential significant risk to our company. The Compensation Committee is responsible for overseeing risks and exposures associated with our compensation programs and arrangements, including our executive and director compensation programs and arrangements, and management succession planning. The Nominating and Corporate Governance Committee oversees risks relating to our corporate governance matters and policies and director succession planning.
Compensation Committee Interlocks
The Compensation Committee is composed entirely of directors who are not our current or former employees, each of whom meets the applicable definition of “independent director” in the current Nasdaq Listing Rules and SEC rules and regulations. None of the members of the Compensation Committee during the fiscal year ended December 31, 2017 was an executive officer of a company of which one of our executive officers is a director. The Compensation Committee is responsible for establishing and administering our executive compensation policies. Our Compensation Committee does not have any interlocks with other companies. Prior to establishing the Compensation Committee, our full Board of Directors made final decisions relating to the compensation of our officers.
EXECUTIVE AND DIRECTOR COMPENSATION
Executive Compensation Overview
The Compensation Committee of our Board of Directors administers our executive compensation programs on behalf of our Board of Directors. The Compensation Committee has a charter that will be reviewed and updated annually, or as may be warranted from time to time. The current members of the Compensation Committee are Michael Giuffre, M.D. (Chair), James Parsons and Richard Pilnik.
This section addresses the compensation of our President and Chief Executive Officer and our only other executive officer as of December 31, 2017:
|
● |
Rick Pauls, our President and Chief Executive Officer; and |
|
● |
Todd Verdoorn, Ph.D., our Chief Scientific Officer. |
The above executive officers are collectively referred to as the named executive officers.
The elements of the compensation program for our named executive officers include:
|
● |
base salary; |
|
● |
long-term equity-based incentive compensation; |
|
● |
annual incentive compensation; and |
|
● |
other compensation, including certain health, welfare and retirement benefits and, when determined necessary, limited perquisites. |
The named executive officers also have termination and change in control benefits as set forth in their respective employment agreements. See “—Post-Termination Severance and Change in Control Arrangements.”
When reading this Executive Compensation Overview, please note that we are an emerging growth company under the JOBS Act and are not required to provide a “Compensation Discussion and Analysis” of the type required by Item 402 of Regulation S-K. This Executive Compensation Overview is intended to supplement the SEC-required disclosure, which is included below this section, and it is not a Compensation Discussion and Analysis.
Base Salary
We provide a base salary for our named executive officers, which, unlike some of the other elements of our executive compensation program, is not subject to company or individual performance risk. We recognize the need for most executives to receive at least a portion of their total compensation in the form of a guaranteed base salary that is paid in cash regularly throughout the year. The base salaries set for our named executive officers are intended to provide a steady income regardless of share price performance, allowing executives to focus on both near-term and long-term goals and objectives without undue reliance on short term share price performance or market fluctuations.
We initially fix base salaries for our executives at a level that we believe enables us to hire and retain them in a competitive environment and to reward satisfactory individual performance and a satisfactory level of contribution to our overall business objectives. The Compensation Committee reviews and approves any increases in base salaries for our named executive officers.
The Compensation Committee has the authority to engage the services of outside experts and advisors as it deems necessary or appropriate to carry out its duties and responsibilities, and prior to doing so, assesses the independence of such experts and advisors from management.
Our Chief Executive Officer assists the Compensation Committee in gathering compensation related data regarding our executive officers and making recommendations to the Compensation Committee regarding the form and amount of compensation to be paid to each executive officer. In addition, the Compensation Committee has retained 21-Group, a compensation consultant, to assist in the design and review of certain aspects of our executive compensation program. The 21-Group does not provide any services to our company other than those for which it has been retained by the Compensation Committee.
In making final decisions regarding compensation to be paid to our executive officers, the Compensation Committee considers the recommendations of our Chief Executive Officer, the data compiled and recommendations of the 21 Group, as well as its own views as to the form and amount of compensation to be paid, the general performance of our company and the individual officers, the performance of our common share price and other factors that may be relevant. Final deliberations and decisions by the Compensation Committee regarding the form and amount of compensation to be paid to our executive officers, including our Chief Executive Officer, are made by the Compensation Committee, without the presence of the Chief Executive Officer or any other executive officer of our company.
Annualized base salary rates for each of our named executive officers for fiscal 2017 and the current fiscal 2018 are as follows:
|
Name |
Fiscal |
Fiscal |
% Change From |
||||||||
|
Rick Pauls |
$ | 280,000 | $ | 345,000 | 23 | ||||||
|
Todd Verdoorn |
200,000 | 240,000 | 20 | ||||||||
Long-Term Equity-Based Incentive Compensation
The long-term equity-based incentive compensation component consists of stock options granted under the DiaMedica Therapeutics Inc. Stock Option Plan (“Stock Option Plan”), which generally vest quarterly over a three-year period and deferred share units (“DSUs”), granted under the DiaMedica Therapeutics Inc. Deferred Share Unit Plan (“DSU Plan”). These plans are designed to give each option and DSU holder an interest in preserving and maximizing shareholder value in the long term, to enable us to attract and retain individuals with experience and ability, and to reward individuals for current performance and expected future performance. Long-term equity-based incentives are intended to comprise a significant portion of each executive’s compensation package, consistent with our executive compensation objective to align the interests of our executives with the interests of our shareholders.
The Compensation Committee uses stock options as a portion of the long-term equity based incentive compensation component since the Compensation Committee believes that options effectively incentivize executives to maximize company performance, as the value of awards is directly tied to an appreciation in the value of our common shares. Stock options also provide an effective retention mechanism because of vesting provisions. An important objective of our long-term equity-based incentive program is to strengthen the relationship between the long-term value of our common shares and the potential financial gain for our executives. Stock options provide recipients with the opportunity to purchase our common shares at a price fixed on the grant date regardless of future market price. Because stock options become valuable only if the share price increases above the exercise price and the option holder remains employed during the period required for the option to vest, they provide an incentive for an executive to remain employed. In addition, stock options link a portion of an executive’s compensation to the interests of our shareholders by providing an incentive to achieve corporate goals and increase the market price of our common shares over the vesting period.
The Compensation Committee previously used DSUs as a portion of the long-term equity-based incentive compensation component in order to provide an alternative form of compensation to satisfy annual and special bonuses payable to our executive officers. The DSU Plan provided that the Board of Directors may, from time to time, issue DSUs to our executive officers at the time of declaring or awarding any bonuses. The number of DSUs granted was determined by dividing the applicable bonus amount by the fair market value of our common shares as of the last trading day before the award date as calculated. No DSUs were granted during 2017 or to date during 2018.
The table below sets forth the stock options that we granted to our named executive officers in 2017 and to date in 2018:
|
Name |
Grant Date |
Number of Shares |
Exercise Price |
||||||
|
Rick Pauls |
06/19/17 |
42,500 | $ | 6.40 | |||||
|
04/17/18 |
33,500 | 11.20 | |||||||
|
Todd Verdoorn |
06/19/17 |
25,000 | 6.40 | ||||||
|
04/17/18 |
21,775 | 11.20 | |||||||
Annual Incentive Compensation
In addition to base salary and long-term equity based incentive compensation, we provide our named executive officers the opportunity to earn annual incentive compensation based on the achievement of certain company and individual related performance goals. Our annual bonus program directly aligns the interests of our executive officers and shareholders by providing an incentive for the achievement of key corporate and individual performance measures that are critical to the success of our company and linking a significant portion of each executive’s annual compensation to the achievement of such measures.
All Other Compensation
It is generally our policy not to extend significant perquisites to our executives that are not available to our employees generally. Our executives receive benefits that are also received by our other employees, including participation in the DiaMedica USA, Inc. 401(k) Plan and health, dental, disability and life insurance benefits.
Employment Agreements
In September 2018, we entered into an employment agreement with each of our executive officers, which provides for an annual base salary, subject to periodic reviews, discretionary bonus and incentive based compensation, equity-based compensation, and benefits, in each case as determined by the Board of Directors (or a committee thereof) from time to time. The agreements contain standard confidentiality, non-competition, non-solicitation and assignment of intellectual property provisions. The agreements also contains standard severance and change in control provisions which are described under “—Post-Termination Severance and Change in Control Arrangements.”
Post Termination Severance and Change in Control Arrangements
Severance Arrangements. Under the terms of the employment agreements with our executive officers, if we terminate the executive’s employment without “cause”, the executive will be entitled to salary continuation payments for 12 months in the case of Mr. Pauls and nine months in the case of each of the other executives, Consolidated Omnibus Budget Reconciliation Act (“COBRA”) premium reimbursement during the salary continuation period, a pro rata portion of his target annual bonus for the year of termination, and immediate acceleration of his equity awards, as severance, subject to executing a separation agreement and release of claims. “Cause” is defined in the employment agreements as: (i) gross negligence or willful failure to perform the executive’s duties and responsibilities to the Company; (ii) commission of any act of fraud, theft, embezzlement, financial dishonesty or any other willful misconduct that has caused or is reasonably expected to result in injury to the Company; (iii) conviction of, or pleading guilty or nolo contendere to, any felony or a lesser crime involving dishonesty or moral turpitude; (iv) material breach by the executive of any of his obligations under the agreement or any written agreement or covenant with the Company, including the policies adopted from time to time by the Company applicable to all executives, that has not been cured within 30 days of notice of such breach; or (v) we terminate the employment of the executive in connection with a liquidation, dissolution or winding down of the Company.
We believe that the form and amount of these severance benefits are fair and reasonable to both the Company and our executives. The Compensation Committee intends to review our severance arrangements periodically to ensure that they remain necessary and appropriate.
Change in Control Arrangements. To encourage continuity, stability and retention when considering the potential disruptive impact of an actual or potential corporate transaction, we have established change in control arrangements, including provisions in our Stock Option Plan and executive employment agreements. These arrangements are designed to incentivize our executives to remain with our company in the event of a change in control or potential change in control.
Under the terms of the employment agreements that we entered into with our executives in September 2018, if we terminate the executive’s employment without “cause” or the executive terminates his employment with “good reason” in connection with or within 12 months after a “change in control,” the executive will be entitled to salary continuation payments for 18 months in the case of Mr. Pauls and 12 months in the case of each of the other executives, COBRA premium reimbursement during the salary continuation period, a pro rata portion of his target annual bonus for the year of termination, and immediate acceleration of his equity awards, as severance, subject to executing a separation agreement and release of claims.
“Good reason” is defined in the employment agreements as the executive’s resignation within 30 days following the expiration of any cure period following the occurrence of one or more of the following, without the executive’s express written consent: (i) a material reduction of the executive’s duties, authority, reporting level, or responsibilities, relative to his duties, authority, reporting level, or responsibilities in effect immediately prior to such change in control; (ii) a material reduction in the executive’s base compensation; or (iii) the Company’s requiring of the executive to change the principal location at which the executive is to perform services by more than 50 miles.
“Change in control” is defined in the employment agreements as the occurrence of any of the following: (i) the acquisition, other than from us, by any individual, entity or group of beneficial ownership of 50% or more of either our then outstanding common shares or the combined voting power of our then outstanding voting securities entitled to vote generally in the election of directors; (ii) the consummation of a reorganization, merger or consolidation of the Company, in each case, with respect to which all or substantially all of the individuals and entities who were the respective beneficial owners of our common shares and voting securities immediately prior to such reorganization, merger or consolidation do not, following such reorganization, merger or consolidation, beneficially own, directly or indirectly, more than 50% of, respectively, of then outstanding common shares and the combined voting power of then outstanding voting securities entitled to vote generally in the election of directors, as the case may be, of the corporation resulting from such reorganization, merger or consolidation; or (iii) the sale or other disposition of all or substantially all of our assets.
We believe these change in control arrangements are an important part of our executive compensation program in part because they mitigate some of the risk for executives working in a smaller company where there is a meaningful risk that the company may be acquired. Change in control benefits are intended to attract and retain qualified executives who, absent these arrangements and in anticipation of a possible change in control of our company, might consider seeking employment alternatives to be less risky than remaining with our company through the transaction. We believe that the form and amount of these change in control benefits are fair and reasonable to both our company and our executives. The Compensation Committee intends to review our change in control arrangements periodically to ensure that they remain necessary and appropriate.
Indemnification Agreements
We have entered into indemnification agreements with all of our executive officers. The indemnification agreements are governed exclusively by and construed according to the substantive laws of the Canada, without regard to conflicts-of-laws principles that would require the application of any other law, and provide, among other things, for indemnification, to the fullest extent permitted by law and our by-laws, against any and all expenses (including attorneys’ fees) and liabilities, judgments, fines and amounts paid in settlement that are paid or incurred by the executive or on his or her behalf in connection with such action, suit or proceeding. We will be obligated to pay these amounts only if the executive acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the best interests of our company and, in the case of a criminal or administrative proceeding that is enforced by a monetary penalty, he or she had reasonable grounds for believing that his or her conduct was lawful. The indemnification agreements provide that the executive will not be indemnified and expenses advanced with respect to an action, suit or proceeding initiated by the executive unless (i) so authorized or consented to by our Board of Directors or the company has joined in such action, suit or proceeding or (ii) the action, suit or proceeding is one to enforce the executive’s rights under the indemnification agreement. Our indemnification and expense advance obligations are subject to the condition that an appropriate person or body not party to the particular action, suit or proceeding shall not have determined that the executive is not permitted to be indemnified under applicable law. The indemnification agreements also set forth procedures that apply in the event an executive requests indemnification or an expense advance.
Summary Compensation Table
The table below provides summary information concerning all compensation awarded to, earned by or paid to our named executive officers during our 2017 and 2016 fiscal years. We did not have any officers during the year ended December 31, 2017, other than Rick Pauls and Todd Verdoorn, Ph.D.
|
Name and Principal Position |
Year |
Salary |
Bonus |
Option Awards(3) |
All Other |
Total |
||||||||||||||||
|
Rick Pauls(1) |
2017 |
$ | 280,000 | $ | 36,667 | $ | 167,738 | $ | 17,550 | $ | 501,956 | |||||||||||
|
President and Chief Executive Officer |
2016 |
276,250 | — | 100,196 | 11,400 | 387,846 | ||||||||||||||||
|
Todd Verdoorn, Ph.D.(2) |
2017 |
200,000 | 40,000 | 98,670 | 7,200 | 345,870 | ||||||||||||||||
|
Chief Scientific Officer |
2016 |
164,792 | — | 58,939 | 4,250 | 227,981 | ||||||||||||||||
______________________________
|
(1) |
Mr. Pauls is also a director of the company and did not receive any compensation related to his role as a director. |
|
(2) |
Dr. Verdoorn became a consultant to the company and was appointed as our Vice President of Neuroscience on January 20, 2016 and became an employee of the company and was promoted to Chief Scientific Officer on May 9, 2016. The portion of his 2016 salary for the period during which he served as a consultant was paid in the form of consulting fees. |
|
(3) |
Amounts reflect the full grant-date fair value of stock options granted during the applicable year computed in accordance with Accounting Standards Codification (ASC) Topic 718, rather than the amounts paid to or realized by the named individual. The grant date fair value is determined based on our Black-Scholes option pricing model. The table below sets forth the specific assumptions used in the valuation of each such option award: |
|
Grant Date |
Grant Date Fair Value Per Share |
Risk Free Interest Rate |
Expected Life |
Expected Volatility |
Expected Dividend Yield |
|||||||||
|
06/19/2017 |
$ | 4.96 | 0.98% |
4.4 years |
119.0% | — | ||||||||
|
11/28/2016 |
3.16 | 1.01% |
5.5 years |
112.5% | — | |||||||||
There can be no assurance that unvested awards will vest (and, absent vesting and exercise, no value will be realized by the executive for the award).
|
(4) |
The amounts shown in the “All Other Compensation” column for fiscal 2017 include the following with respect to each named executive officer: |
|
Name |
401(k) Match |
Health Savings Account Contribution |
Total |
|||||||||
|
Rick Pauls |
$ | 10,800 | $ | 6,750 | $ | 17,550 | ||||||
|
Todd Verdoorn, Ph.D. |
7,200 | — | 7,200 | |||||||||
Outstanding Equity Awards at Fiscal Year-End
The following table presents for each named executive officer information regarding outstanding equity awards held as of December 31, 2017.
|
Option Awards |
Stock Awards |
||||||||||||||||||||
|
Name |
Number of Securities Underlying Unexercised Options (#) Exercisable |
Number of Securities Underlying Unexercised Options (#) Unexercisable(1) |
Option Exercise Price CAD($) |
Option Expiration Date(2) |
Number of Shares or Units of Stock That Have Not Vested(3) |
Market Value of Shares or Units of Stock That Have Not Vested(4) |
|||||||||||||||
|
Rick Pauls |
|||||||||||||||||||||
|
Stock Options |
10,000 | — | $ | 23.00 |
10/06/2021 |
||||||||||||||||
| 10,000 | — | 34.00 |
02/15/2022 |
||||||||||||||||||
| 10,000 | — | 21.40 |
06/25/2023 |
||||||||||||||||||
| 45,000 | 22,500 | 3.00 |
12/01/2025 |
||||||||||||||||||
| 14,167 | 28,333 | 5.20 |
11/28/2026 |
||||||||||||||||||
| 7,083 | 35,417 | 6.40 |
06/19/2027 |
||||||||||||||||||
|
DSUs |
1,749 | $ | 8,069 | ||||||||||||||||||
|
Todd Verdoorn, Ph.D. |
|||||||||||||||||||||
|
Stock Options |
4,800 | 2,400 | 3.00 |
12/01/2025 |
|||||||||||||||||
| 8,333 | 16,667 | 5.20 |
11/28/2026 |
||||||||||||||||||
| 4,167 | 20,833 | 6.40 |
06/19/2027 |
||||||||||||||||||
|
DSUs |
— | — | |||||||||||||||||||
_____________________________
|
(1) |
All stock options vest in 12 equal quarterly installments over three years. |
|
(2) |
All stock options have a 10-year term, but may terminate earlier if the recipient’s employment or service relationship with our company terminates. |
|
(3) |
All DSU awards are settled after the recipient’s employment or service relationship with our company terminates. |
|
(4) |
The market value of DSU awards that have not been settled as of December 31, 2017 is based on the closing sale price of our common shares as reported by the TSX Venture Exchange on the last trading day of our fiscal year, December 29, 2017 (CAD$ to US$ fixed rate $0.7953). |
Employee Benefit and Stock Plans
Stock Option Plan
The DiaMedica Therapeutics Inc. Amended and Restated Stock Option Plan was adopted by the Board of Directors on September 30, 2018 and by our shareholders on November 6, 2018.
Shares Available. The number of common shares reserved for issuance under the Option Plan at any time is equal to the lesser of: 783,918 (subject to adjustment) and 10% of the issued common shares at the relevant time and the aggregate number of common shares reserved for issuance under any other compensation or incentive mechanism or plan (including deferred share unit plans or employee stock option plans, if any), shall not exceed 10% of our issued shares at the relevant time. In addition, the maximum number of common shares that may be issued under Option Plan upon the exercise of incentive stock options within the meaning of Section 422 of the Code is 283,918 shares (subject to adjustment).
The Option Plan also provides that the number of common shares reserved for issuance:
|
● |
to any one person, within any 12 month period, will not exceed 5% of the issued and outstanding common shares at the time of the grant; |
|
● |
to any one consultant, within any 12 month period, will not exceed 2% of the issued and outstanding common shares at the time of the grant; and |
|
● |
in aggregate to insiders will not exceed 10% of the issued and outstanding common shares at the time of the grant and in aggregate will not exceed, within any 12 month period, 10% of the issued and outstanding common shares at the time of the grant. |
Eligible Participants. Directors, officers, employees and certain consultants of DiaMedica and our subsidiaries are eligible to participate in the Option Plan. Only employees are eligible to receive incentive stock options. No options may be granted to a consultant that provides services (a) in connection with the offer and sale of our securities in a capital raising transaction or (b) which directly or indirectly promote or maintain a market for our securities.
Awards Available. The Option Plan authorizes the award of stock options, including incentive stock options within the meaning of Section 422 of the Code. Options will have an expiry date not exceeding 10 years from the date of grant, after which they cease to be exercisable. Subject to the conditions in the Option Plan, the Board of Directors determines the manner in which an option shall vest and become exercisable.
Transferability. Options are exercisable only by the participant to whom they are granted and may not be assigned or transferred. Notwithstanding this restriction, upon the death of a participant, the participant's legal representatives, heirs, executors and administrators may exercise the participant's options for a period ending no later than the earlier of the option expiry date and 12 months after the participant's death.
Effect of Termination of Employment or Service. Subject to the discretion of the Board of Directors, where a person ceases to be an eligible participant under the Option Plan, other than by reason of death or in the event of termination for cause, Options granted to participants will cease to be exercisable on the earlier of the expiry date and 90 days after the date of termination. Subject to the discretion of the Board of Directors, if a participant is terminated for cause, all Options received will terminate and cease to be exercisable upon such termination.
Certain Adjustments. In the event of any change in our outstanding common shares by reason of any stock dividend, split, recapitalization, reclassification, amalgamation, merger, consolidation, combination or exchange of shares or distribution of rights to holders of shares or any other form of corporate reorganization whatsoever, an equitable adjustment will be made to the share limits in the Option Plan and any Options then outstanding and the exercise price in respect of such Options.
Termination/Amendment. The Option Plan will terminate on November 5, 2028 and may be terminated prior to such time by the Board of Directors. No Options will be granted after termination of the Option Plan, but Options outstanding will remain outstanding in accordance with their applicable terms and conditions and the terms and conditions of the Option Plan. Subject to limitations contained in the Option Plan, the Board of Directors may amend, modify or terminate the Option Plan.
Plan Administration. Although the Option Plan is administered by the Compensation Committee, the Board of Directors must make all grants of Options under the Option Plan.
Deferred Share Unit Plan
The DiaMedica Therapeutics Inc. Deferred Share Unit Plan was adopted by the Board of Directors on August 25, 2011 and by our shareholders on September 22, 2011.
Shares Available. The number of common shares reserved for issuance under the DSU Plan at any time is 100,000 shares (subject to adjustment). In no event may the number of common shares reserved for issuance to any one person pursuant to DSUs and options exceed 5% of our outstanding common shares. The DSU Plan also provides that the number of common shares reserved for issuance in aggregate to insiders will not exceed 10% of the issued and outstanding common shares at the time of the grant and in aggregate will not exceed, within any 12 month period, 10% of the issued and outstanding common shares at the time of the grant.
Eligible Participants. Directors and executive officers of DiaMedica and our subsidiaries are eligible to participate in the DSU Plan.
Awards Available. The DSU Plan authorizes the award of deferred share units, which is a right to receive, on a deferred payment basis, a common share or the fair market value thereof, or a combination thereof. At the time of grant, the Board of Directors decides the total compensation that will be satisfied in the form of DSUs.
Transferability. DSUs and all other rights, benefits or interests in the DSU Plan are non-transferable.
Effect of Termination of Service. A holder of a DSU who has terminated his or her employment or service with DiaMedica may elect to receive one common share with respect to each whole DSU credit to his or her account, net of required tax withholding obligations, by filing a notice of redemption on or before December 15th of the first calendar year commencing after the date on which the holder’s employment or service has terminated. In the event of the death of a holder of a DSU, we will within two months of such death pay cash equal to the fair market value of the common shares that would have otherwise been issued upon a termination of employment or service.
Certain Adjustments. In the event of any dividend paid in shares, share subdivision, combination or exchange of shares, merger, consolidation, spin-off or other distribution of DiaMedica assets to shareholders, or any other change in our capital affecting our common shares, the Board will make with respect to the number of DSUs outstanding under the DSU Plan, any proportionate adjustments as it considers appropriate to reflect that change.
Termination/Amendment. The DSU Plan may be terminated by the Board of Directors at any time. No DSUs will be granted after termination of the DSU Plan, but DSUs outstanding will remain outstanding in accordance with their applicable terms and conditions and the terms and conditions of the DSU Plan. Subject to limitations contained in the DSU Plan, the Board of Directors may amend, modify or terminate the DSU Plan.
Plan Administration. Although the DSU Plan is administered by the Compensation Committee, the Board of Directors must make all grants of DSUs under the DSU Plan.
Non-Employee Director Compensation
The table below provides summary information concerning the compensation of each individual who served as a director of our company during the fiscal year ended December 31, 2017, other than Rick Pauls, our President and Chief Executive Officer, who was not compensated separately for serving on the Board of Directors during fiscal 2017. His compensation during fiscal 2017 for serving as an executive officer of our company is set forth under “—Summary Compensation Table.”
|
Name |
Fees Earned or Paid in Cash |
Option Awards(1) |
Stock Awards |
All Other Compensation ($) |
Total |
|||||||||||||||
|
Michael Giuffre, M.D. |
$ | 15,906 | $ | 19,723 | — | — | $ | 35,629 | ||||||||||||
|
James Parsons |
15,906 | 19,723 | — | — | 35,629 | |||||||||||||||
|
Richard Pilnik |
31,812 | 19,723 | — | — | 51,535 | |||||||||||||||
|
Zhenyu Xiao |
15,906 | 19,723 | — | — | 35,629 | |||||||||||||||
_______________________________
|
(1) |
On June 19, 2017, each non-employee director received a stock option to purchase a 5,000 common shares at an exercise price of CAD$6.40 per share granted under our Stock Option Plan. Such option expires on June 19, 2027 and vests in 12 equal quarterly installments over three years. The amounts reflected represent the grant date fair value for option awards granted to each non-employee director computed in accordance with FASB ASC Topic 718. |
We use a combination of retainer fees and long-term equity-based incentive compensation in the form of stock option grants to attract and retain qualified candidates to serve on the Board of Directors. For fiscal 2017, each of our non-employee directors earned annual retainers and meeting fees. Each non-employee director earned a $13,918 annual retainer and the Chair of our Audit Committee and Compensation Committee earned an additional $1,988 annual retainer. The Chairman of the Board earned an additional $15,906. The annual retainers were accrued and unpaid as of December 31, 2017. All of our directors are reimbursed for travel expenses for attending meetings and other miscellaneous out-of-pocket expenses incurred in performing their Board functions.
For the reasons noted above, long-term equity based incentive compensation is a significant component of how we compensate directors. Directors generally receive annual grants with a fair market value equivalent to their cash compensation. These grants vest in 12 equal quarterly installments over three years and expire on the tenth anniversary of the grant date.
Limitation of Liability and Indemnification Matters
Our by-laws provide that no director or officer will be liable for the acts, receipts, neglects or defaults of any other director or officer or employee, or for joining in any receipt or other act for conformity, or for any loss, damage or expense happening to us through the insufficiency or deficiency of title to any property acquired for or on behalf of us, or for the insufficiency or deficiency of any security in or upon which any of our moneys will be invested, or for any loss or damage arising from the bankruptcy, insolvency or tortious acts of any person with whom any of our moneys, securities or effects are deposited, or for any other loss, damage or misfortune whatever which will happen in the execution of the duties of his office or in relation thereto, unless the same are occasioned by his own willful neglect or default; provided that such provision will not relieve any director or officer from the duty to act in accordance with applicable corporate law or from liability for any breach thereof.
Our by-laws provide that subject to certain limitations, we will indemnify a director or officer, a former director or officer, or a person who acts or acted at our request as a director or officer of a body corporate of which we are or was a shareholder or creditor (or a person who undertakes or has undertaken any liability on behalf of us or any such body corporate) and his heirs and legal representatives, against any and all costs, charges and expenses, including an amount paid to settle an action or satisfy a judgment, reasonably incurred by him in respect of any civil, criminal or administrative action or proceeding to which he is made a party by reason of being or having been a director or officer, if: (1) the officer or director acted honestly and in good faith with a view to the best interests of our company; and (2) in the case of a criminal or administrative action or proceeding that is enforced by a monetary penalty, the officer or director has reasonable grounds for believing that his or her conduct was lawful. Subject to applicable law and the approval of the Board of Directors, we may advance anticipated defense costs in respect of the foregoing.
We entered into indemnification agreements with all of our directors, which are nearly identical to the indemnification agreements with our executive officers as described under “—Executive Compensation Overview—Indemnification Agreements.”
At present, there is no pending litigation or proceeding involving any of our directors or executive officers as to which indemnification is required or permitted, and we are not aware of any threatened litigation or proceeding that may result in a claim for indemnification.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, executive officers or persons controlling us, we have been informed that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
Related Person Relationships and Transactions
Other than as described below or under the heading “Executive and Director Compensation,” we have not identified any transactions since January 1, 2016 to which we have been a party, in which the amount involved exceeded or will exceed the lesser of $120,000 or 1% of the average of our total assets at year end for the last two fiscal years, and in which any of our executive officers, directors or holders of more than 5% of our common shares, or an affiliate or immediate family member thereof, had or will have a direct or indirect material interest.
Participation in Private Placement
On March 29, 2018, we completed, in two tranches, a brokered and non-brokered private placement of 1,322,965 units at a price of $4.90 per unit for aggregate gross proceeds of approximately $6.3 million. Each unit consisted of one common share and one-half of one common share purchase warrant. Each whole warrant entitles the holder to purchase one common share at a price of $7.00 at any time prior to expiration on March 19, 2020 and March 29, 2020 for tranche 1 and tranche 2, respectively.
Rick Pauls, our President and Chief Executive Officer and a member of our Board of Directors, Scott Kellen, our Chief Financial Officer, and Michael Giuffre, M.D., a member of our Board of Directors, each participated in the offering on the same terms and conditions as other investors, as set forth in the table below:
|
Name |
Purchase Price |
Number of Common Shares |
Number of Common Shares Underlying Warrants |
||||||||||
|
Rick Pauls |
$ | 20,090 | 4,100 | 2,050 | |||||||||
|
Scott Kellen |
10,000 | 2,040 | 1,020 | ||||||||||
|
Michael Giuffre, M.D. |
110,000 | 22,449 | 11,225 | ||||||||||
|
Total |
$ | 140,090 | 28,589 | 14,295 | |||||||||
Relationship with Hermeda Industrial Co., Limited
We and Hermeda Industrial Co., Limited (“Hermeda”) are parties to an investment agreement, which includes terms relating to the composition of our Board of Directors. Under director nomination provisions of this agreement, Hermeda has the right to designate a representative to be nominated to our Board of Directors for so long as Hermeda beneficially owns at least 10% of our outstanding common shares on a non-diluted basis, and we agreed to use our reasonable best efforts to cause the Hermeda designee to be elected. As of November 15, 2018, Hermeda beneficially owned 12.7% of our outstanding common shares. Zhenyu Xiao, Ph.D., one of our directors, is the Director of Hermeda and is the current designee of Hermeda under the investment agreement. In the event Hermeda has no representative on our Board of Directors and beneficially owns at least 10% of our outstanding common shares, on a non-diluted basis, and provides notice to us of its representative, we shall take such steps that are necessary for our Board of Directors to appoint the representative as a member of our Board of Directors.
To induce Hermeda to enter into the investment agreement, two members of our Board of Directors, Rick Pauls and Michael Giuffre, M.D., and certain of their related parties entered into voting agreements with DiaMedica pursuant to which these individuals agreed to vote their DiaMedica common shares in favor of the Hermeda designee to the Board of Directors at the then next annual general meeting of shareholders.
License Agreement
In September 2018, we entered into a license and collaboration agreement with Ahon Pharma, a subsidiary of Fosun Pharma, which allows Ahon Pharma to have exclusive rights to develop and commercialize DM199 for acute ischemic stroke in mainland China, Taiwan, Hong Kong S.A.R. and Macau S.A.R. Under the terms of the agreement, we are entitled to receive an upfront payment of $5.0 million, consisting of $500,000 on signing and $4.5 million upon regulatory clearance to initiate a clinical trial in China. We also have the potential to receive an additional $27.5 million in development and sales related milestones and up to approximately 10% royalties on net sales of DM199 in the licensed territories. All development, regulatory, sales, marketing, and commercial activities and associated costs in the licensed territories will be the sole responsibility of Ahon Pharma. This agreement may be terminated at any time by Ahon Pharma by providing 120 days written notice. Fosun Pharma, through its partnership with SK Group, a South Korea based company is an investor in DiaMedica through its equity investment in 2016.
Indemnification Agreements
We have entered into indemnification agreements with all of our directors and executive officers. The indemnification agreements provide, among other things, for indemnification, to the fullest extent permitted by law and our by-laws, against any and all expenses (including attorneys’ fees) and liabilities, judgments, fines and amounts paid in settlement that are paid or incurred by the executive or on his or her behalf in connection with such action, suit or proceeding. The indemnification agreements also set forth procedures that apply in the event an executive requests indemnification or an expense advance. For more information regarding these agreements, see “Executive and Director Compensation—Limitations on Liability and Indemnification Matters.”
Policies and Procedures for Related Party Transactions
The Board of Directors has delegated to the Audit Committee, pursuant to the terms of a written policy, the authority to review, approve and ratify related party transactions. If it is not feasible for the Audit Committee to take an action with respect to a proposed related party transaction, the Board of Directors or another committee, may approve or ratify it. No member of the Board of Directors or any committee may participate in any review, consideration or approval of any related party transaction with respect to which such member or any of his or her immediate family members is the related party.
Our policy defines a “related party transaction” as a transaction, arrangement or relationship (or any series of similar transactions, arrangements or relationships) in which we (including any of our subsidiaries) were, are or will be a participant and in which any related party had, has or will have a direct or indirect interest.
Prior to entering into or amending any related party transaction, the party involved must provide notice to our finance department of the facts and circumstances of the proposed transaction, including:
|
● |
the related party’s relationship to us and his or her interest in the transaction; |
|
● |
the material facts of the proposed related party transaction, including the proposed aggregate value of such transaction or, in the case of indebtedness, the amount of principal that would be involved; |
|
● |
the purpose and benefits of the proposed related party transaction with respect to us; |
|
● |
if applicable, the availability of other sources of comparable products or services; and |
|
● |
an assessment of whether the proposed related party transaction is on terms that are comparable to the terms available to an unrelated third party or to employees generally. |
If the finance department determines the proposed transaction is a related party transaction, the proposed transaction will be submitted to the Audit Committee for consideration. In determining whether to approve a proposed related party transaction, the Audit Committee will consider, among other things, the following:
|
● |
the purpose of the transaction; |
|
● |
the benefits of the transaction to us; |
|
● |
the impact on a director’s independence in the event the related party is a non-employee director, an immediate family member of a non-employee director or an entity in which a non-employee director is a partner, shareholder or executive officer; |
|
● |
the availability of other sources for comparable products or services; |
|
● |
the terms of the transaction; and |
|
● |
the terms available to unrelated third parties or to employees generally. |
Under our policy, certain related party transactions as defined under our policy will be deemed to be pre-approved by the Audit Committee and will not be subject to these procedures.
PRINCIPAL SHAREHOLDERS
The following table sets forth information known to us with respect to the beneficial ownership of our common shares as of November 15, 2018 for:
|
● |
each person known by us to beneficially own more than five percent of the outstanding shares of our common shares; |
|
● |
each of our directors; |
|
● |
each of the executive officers named in the Summary Compensation Table included earlier in this prospectus under the heading “Executive and Director Compensation;” and |
|
● |
all of our current directors and executive officers as a group. |
Ownership information provided below assumes no exercise of the underwriter’s over-allotment option.
The columns entitled “Shares Beneficially Owned” and “Percentage of Common Shares Beneficially Owned Prior to Offering” are based on common shares outstanding as of , 2018. The column entitled “Percentage of Common Shares Beneficially Owned After Offering” is based on common shares to be outstanding after this offering, after giving effect to the sale of common shares in this offering. The table below does not reflect any common shares that those listed in the table may purchase in this offering.
Beneficial ownership is determined according to the rules of the SEC and generally means that a person has beneficial ownership of a security if he or she possesses sole or shared voting or investment power of that security, including the right to acquire beneficial ownership of that security within 60 days, including through outstanding options and warrants that are exercisable within 60 days of , 2018. Options and warrants to purchase common shares that are exercisable within 60 days of , 2018 are deemed to be beneficially owned by the persons possessing those rights and are treated as outstanding for the purpose of computing percentage ownership of that person, but are not treated as outstanding for the purpose of computing any other person’s ownership percentage. Except as indicated in the footnotes below, each of the beneficial owners named in the table below has, and upon completion of this offering will have, to our knowledge, sole voting and investment power with respect to all common shares listed as beneficially owned by him or her, except for shares owned jointly with that person’s spouse. Unless otherwise indicated, the address for each of the shareholders in the table below is DiaMedica Therapeutics Inc., 2 Carlson Parkway, Suite 260, Minneapolis, MN 55447.
|
Title of Class |
|
Name and Address of Beneficial Owner |
|
Amount and Nature of |
|
Percent of Class |
|||
|
Directors and Officers: |
|
|
|
|
|
Prior to Offering |
|
After Offering |
|
|
Common Shares |
|
Richard Pilnik |
|
57,917 |
|
* |
|
|
|
|
Common Shares |
|
Michael Giuffre, M.D. |
|
186,637 |
(2) |
|
2.4% |
|
|
|
Common Shares |
|
James Parsons |
|
18,333 |
|
* |
|
|
|
|
Common Shares |
|
Zhenyu Xiao, Ph.D. |
|
1,003,000 |
(3) |
|
12.8% |
|
|
|
Common Shares |
|
Rick Pauls |
|
177,772 |
|
2.2% |
|
|
|
|
Common Shares |
|
Todd Verdoorn |
|
40,996 |
|
* |
|
|
|
|
Common Shares |
|
All current directors and executive officers as a group (8 persons) |
|
1,498,173 |
|
18.3% |
|
|
|
|
Significant Beneficial Owners: |
|
|
|
|
|
|
|
|
|
|
Common Shares |
|
Hermeda Industrial Co., Limited |
|
1,000,000 |
(3) |
|
12.7% |
|
|
|
Common Shares |
|
CentreStone Ventures, LP |
|
705,917 |
(4) |
|
9.0% |
|
|
|
Common Shares |
|
Nancy Chang |
|
657,895 |
(5) |
|
8.4% |
|
|
_________________________
* Represents beneficial ownership of less than one percent.
|
(1) |
Includes for the persons listed below the following common shares subject to options and warrants held by such persons that are currently exercisable or become exercisable within 60 days of November 15, 2018: |
|
Name |
Common Shares Underlying |
Common Shares Underlying |
||||
|
Directors |
||||||
|
Richard Pilnik |
47,917 | — | ||||
|
Michael Giuffre, M.D. |
24,583 | 11,225 | ||||
|
James Parsons |
18,333 | — | ||||
|
Zhenyu Xiao, Ph.D. |
3,000 | — | ||||
|
Named Executive Officers |
||||||
|
Rick Pauls |
152,667 | 2,050 | ||||
|
Todd Verdoorn |
39,996 | — | ||||
|
All current directors and executive officers as a group (8 persons) |
296,954 | 14,295 |
Excludes common shares issuable upon the settlement of DSUs held by: Pilnik (7,588 common shares); Giuffre (4,146 common shares); Parsons (3,850 common shares); Xiao (3,850 common shares); and Pauls (1,749 common shares).
|
(2) |
Includes: (i) 5,165 common shares held by 424822 Alberta Ltd, Michael Giuffre, M.D. has sole voting and dispositive power over the common shares held by 424822 Alberta Ltd., (ii) 36,498 common shares Dr. Giuffre and his wife hold jointly, (iii) 54,186 common shares held by Dr. Giuffre’s sons and daughters, (iv) 21,070 common shares held by Dr. Giuffre’s wife and (v) 33,910 common shares held directly by Dr. Giuffre. |
|
(3) |
Includes 1,000,000 common shares held by Hermeda Industrial Co., Limited. Zhenyu Xiao, Ph.D. is the Director of Hermeda Industrial Co., Limited and has sole voting and dispositive power over the common shares held by Hermeda Industrial Co., Limited. |
|
(4) |
Albert D. Friesen, the managing director of CentreStone Ventures, Inc., has sole voting and dispositive power over the common shares held by CentreStone Ventures, LP. |
|
(5) |
Includes 39,470 shares held by the Chang Family Foundation. Nancy Chang has sole voting and dispositive power over the common shares held by Chang Family Foundation. Also includes 2,500 common shares subject to an option that is currently exercisable or becomes exercisable within 60 days of November 15, 2018. |
DESCRIPTION OF SHARE CAPITAL
We have an authorized share capital consisting of an unlimited number of voting common shares, no par value per share. As of November 15, 2018, there were 7,856,876 voting common shares issued and outstanding. The following description summarizes the most important terms of our common shares. Because it is only a summary, it does not contain all the information that may be important to you. For a complete description, you should refer to our articles and by-laws, which are included as exhibits to the registration statement of which this prospectus forms a part, and to the applicable provisions of the Canada Business Corporation Act (“CBCA”).
Certain Rights of the Common Shares
Dividends
Holders of our voting common shares are entitled to share pro rata in such dividends as may be declared by our Board of Directors. Pursuant to the provisions of the CBCA, we may not declare or pay a dividend if there are reasonable grounds for believing that (1) we are, or would after the payment be, unable to pay our liabilities as they become due or (2) the realizable value of our assets would thereby be less than the aggregate of our liabilities and stated capital of all classes. We may pay a dividend by issuing fully paid shares, or in money or property.
Liquidation, Dissolution or Winding-Up
In the event of a voluntary or involuntary liquidation, dissolution or winding up of the Company or any other distribution of our assets among our shareholders for the purpose of winding-up our affairs, holders of voting common shares are entitled to share pro rata in our assets available for distribution after we pay our creditors.
Voting Rights and Shareholders’ Meetings
Holders of our voting common shares are entitled to receive notice of and to attend and vote at all meetings of our shareholders. Each holder of our voting common shares is entitled to one vote, either in person or by proxy, on all matters submitted to shareholders.
Our Board of Directors must call an annual meeting of shareholders to be held not later than 15 months after the last preceding annual meeting of shareholders but no later than six months after the end of our preceding financial year end and may, at any time, call a special meeting of shareholders. Under our articles, a meeting of our shareholders may be held anywhere in or outside of Canada. For purposes of determining the shareholders who are entitled to receive notice of or to vote at a meeting of shareholders, the Board of Directors may, in accordance with National Instrument 54-101 - Communications with Beneficial Owners of Securities of a Reporting Issuer of the Canadian Securities Administrators, fix in advance a date as the record date for that determination of shareholders, but that record date may not be more than 60 days or less than 30 days before the date on which the meeting is to be held.
The CBCA provides that notice of the time and place of a meeting of shareholders must be sent to each shareholder entitled to vote at the meeting, each director and to our auditors, not more than 60 days and not less than 21 days prior to the meeting. Under our by-laws, the presence at a shareholder meeting, in person or represented by proxy, of at least two shareholders holding not less than one-third of the outstanding voting common shares shall constitute a quorum for the purpose of transacting business at the shareholder meeting. A shareholder may participate in a meeting by means of telephone or other communication facilities that permit all persons participating in the meeting to communicate adequately with each other during the meeting
In the case of joint shareholders, one of the holders present at a meeting may, in the absence of the other holder(s) of the shares, vote the shares. If two or more joint shareholders are present in person or by proxy, then they are to vote as one on the shares held jointly by them.
No Preemption Rights; Limited Restrictions on Directors’ Authority to Issue Common Shares
Existing holders of our voting common shares have no rights of preemption or first refusal under our articles, by-laws or the CBCA with respect to future issuances of our voting common shares. The voting common shares do not have conversion rights, are not subject to redemption and do not have the benefit of any sinking fund provisions. Subject to the rules and policies of The Nasdaq Stock Market and the TSX Venture Exchange and applicable corporate and securities laws, our Board of Directors has the authority to issue additional voting common shares.
Amendments to our Articles and By-laws
Our articles, our by-laws and the CBCA govern the rights of holders of our shares.
Our shareholders can authorize the alteration of our articles to create additional classes of shares or to vary the rights or restrictions attached to any class of our shares by passing a special resolution approved by the holders of at least two-thirds of each class of affected shares represented in person or by proxy at a duly convened meeting of shareholders. Such a special resolution will not be effective until articles of amendment are filed with the Director appointed pursuant to the CBCA.
Our Board of Directors may, by resolution, make, amend or repeal any by-laws that regulate our business or affairs; provided that the Board of Directors shall submit a by-law, or an amendment or a repeal of a by-law, to the shareholders at the next meeting of the shareholders, and the shareholders may, by ordinary resolution, confirm, reject or amend the by-law, amendment or repeal. A by-law, or an amendment or a repeal of a by-law, is effective from the date of the resolution of the Board of Directors until it is confirmed, confirmed as amended or rejected by the shareholders.
Fundamental Changes
Pursuant to the CBCA, we may not effect any of the following fundamental changes without the consent of the holders of at least two-thirds of each class of our outstanding shares represented in person or by proxy and voting separately as a class at a duly convened meeting of our shareholders:
|
● |
any proposed amalgamation involving our company in respect of which the CBCA requires that the approval of our shareholders be obtained; |
|
● |
any proposed plan of arrangement pursuant to the CBCA involving our company in respect of which the CBCA or any order issued by an applicable court requires that the approval of our shareholders be obtained; |
|
● |
any proposed sale, lease or exchange of all or substantially all our assets or property; and |
|
● |
any dissolution, liquidation or winding-up of our company. |
Election and Removal of Directors
At each annual meeting of shareholders, our shareholders are required to elect directors to hold office for a term expiring not later than the close of the next annual meeting of shareholders. Our Board of Directors may fill vacancies among the Board. Our directors may also, between annual meetings of our shareholders, appoint one or more additional directors to serve until the next annual meeting of shareholders; provided, however, that the number of additional directors shall not at any time exceed one-third (1/3) of the number of directors who held office at the expiration of the last meeting of shareholders.
Since shareholders do not have cumulative voting rights, holders of more than 50% of our outstanding common shares can elect all of our directors if they choose to do so. In such event, holders of the remaining shares will be unable to elect any director.
Under the CBCA, at least 25% of our directors must be resident Canadians.
Options, Deferred Share Units and Warrants
Options
As of November 15, 2018, we had outstanding options to purchase an aggregate of 639,359 common shares, with a weighted-average exercise price of $7.87 per share.
Deferred Share Units
As of November 15, 2018, we had outstanding deferred share units which will be converted into 21,183 common shares.
Warrants
As of November 15, 2018, we had outstanding warrants to purchase an aggregate of 807,563 common shares, with a weighted-average exercise price of $6.67 per share.
Registration Rights
We have not granted any rights to have common shares or other securities registered under the Securities Act.
Anti-takeover Laws
In Canada, takeover bids are governed by provincial corporate and securities laws and the rules of applicable stock exchanges. The following description of the rules relating to acquisitions of securities and takeover bids to which Canadian corporate and securities laws apply does not purport to be complete and is subject, and qualified in its entirety by reference, to applicable corporate and securities laws, which may vary from province to province.
A party (the “acquiror”) who acquires beneficial ownership of, or control or direction over, more than 10% of the voting or equity securities of any class of a reporting issuer (or securities convertible into voting or equity securities of any class of a reporting issuer) will generally be required to file with applicable provincial regulatory authorities both a news release and a report containing the information prescribed by applicable securities laws. Subject to the below, the acquiror (including any party acting jointly or in concert with the acquiror) will be prohibited from purchasing any additional securities of the class of the target company previously acquired for a period commencing on the occurrence of an event triggering the aforementioned filing requirement and ending on the expiry of one business day following the filing of the report. This filing process and the associated restriction on further purchases also apply in respect of subsequent acquisitions of 2% or more of the securities of the same class (or securities convertible into voting or equity securities of any class of a reporting issuer). The restriction on further purchases does not apply to an acquiror that beneficially owns, or controls or directs, 20% or more of the outstanding securities of that class.
In addition to the foregoing, certain other Canadian legislation may limit a Canadian or non-Canadian entity’s ability to acquire control over or a significant interest in us, including the Competition Act (Canada) and the Investment Canada Act (Canada). Issuers may also approve and adopt shareholder rights plans or other defensive tactics designed to be triggered upon the commencement of an unsolicited bid and make the company a less desirable takeover target.
Shareholder Rights Plan
We adopted a shareholder rights plan agreement (the “Rights Plan”). The Rights Plan is designed to provide adequate time for the Board of Directors and the shareholders to assess an unsolicited takeover bid for DiaMedica, to provide the Board of Directors with sufficient time to explore and develop alternatives for maximizing shareholder value if a takeover bid is made, and to provide shareholders with an equal opportunity to participate in a takeover bid and receive full and fair value for their common shares. The Rights Plan was renewed at the Company’s annual meeting of shareholders in December 2017 and is set to expire at the close of the Company’s annual meeting of shareholders in 2020.
The rights issued under the Rights Plan will initially attach to and trade with the common shares and no separate certificates will be issued unless an event triggering these rights occurs. The rights will become exercisable only when a person, including any party related to it, acquires or attempts to acquire 20% or more of the outstanding common shares without complying with the “Permitted Bid” provisions of the Rights Plan or without approval of the Board of Directors. Should such an acquisition occur or be announced, each right would, upon exercise, entitle a rights holder, other than the acquiring person and related persons, to purchase common shares at a 50% discount to the market price at the time.
Under the Rights Plan, a Permitted Bid is a bid made to all holders of the common shares and which is open for acceptance for not less than 60 days. If at the end of 60 days at least 50% of the outstanding common shares, other than those owned by the offeror and certain related parties have been tendered, the offeror may take up and pay for the common shares but must extend the bid for a further 10 days to allow other shareholders to tender.
The issuance of common shares upon the exercise of the rights is subject to receipt of certain regulatory approvals.
Listing; Exchange, Transfer Agent and Registrar
Our common shares trade in Canada on the TSX Venture Exchange under the trading symbol “DMA” and over-the-counter in the United States on the OTCQB marketplace under the trading symbol “DMCAF.” We have applied to list our common shares on The Nasdaq Capital Market under the trading symbol “DMAC.”
The transfer agent and registrar for our common shares is Computershare Trust Company.
Other Canadian Laws Affecting U.S. Shareholders
There are no governmental laws, decrees or regulations in Canada relating to restrictions on the export or import of capital, or affecting the remittance of interest, dividends or other payments by us to non-residents of Canada.
Dividends paid by the Company to residents of the United States of America within the meaning of the Canada-United States Tax Convention (1980) (the “US Treaty”) are generally subject to a 15% withholding tax on the gross amount of the dividends (or a 5% withholding tax if the beneficial shareholder is a company which owns at least 10% of the outstanding voting common shares of the Company) pursuant to Article X of the US Treaty. Dividends paid by the Company to other non-residents of Canada are subject to a 25% withholding tax on the amount of the dividends, unless reduced by an applicable tax treaty.
There are no limitations specific to the rights of non-residents of Canada to hold or vote our common shares under the federal laws of Canada, or in our articles or by-laws, other than those imposed by the Investment Canada Act (Canada) as discussed below.
Non-Canadian investors who acquire a controlling interest in us may be subject to the Investment Canada Act (Canada), which governs the basis on which non-Canadians may invest in Canadian businesses. Under the Investment Canada Act (Canada), the acquisition of a majority of the voting interests of an entity (or of a majority of the undivided ownership interests in the voting common shares of an entity that is a corporation) is deemed to be an acquisition of control of that entity. The acquisition of less than a majority but one-third or more of the voting common shares of a corporation (or of an equivalent undivided ownership interest in the voting common shares of the corporation) is presumed to be acquisition of control of that corporation unless it can be established that, on the acquisition, the corporation is not controlled in fact by the acquirer through the ownership of the voting common shares. The acquisition of less than one-third of the voting common shares of a corporation (or of an equivalent undivided ownership interest in the voting common shares of the corporation) is deemed not to be acquisition of control of that corporation.
Differences in Corporate Law
We are governed by the CBCA, which is generally similar to laws applicable to United States corporations. Significant differences between the CBCA and the Delaware General Corporate Law (“DGCL”), which governs companies incorporated in the State of Delaware, include the following:
|
Capital Structure |
||
|
Delaware |
Canada |
|
|
Under the DGCL, the certificate of incorporation must set forth the total number of shares of stock which the corporation shall have authority to issue and the par value of each of such shares, or a statement that the shares are to be without par value. |
Under the CBCA, the articles of incorporation may but are not required to set forth the maximum number of shares that the corporation is authorized to issue. |
|
|
Dividends |
||
| Delaware | Canada | |
|
The DGCL generally provides that, subject to certain restrictions, the directors of a corporation may declare and pay dividends upon the shares of its capital stock either out of the corporation’s surplus or, if there is no such surplus, out of its net profits for the fiscal year in which the dividend is declared and/or the preceding fiscal year. Further, the holders of preferred or special stock of any class or series may be entitled to receive dividends at such rates, on such conditions and at such times as stated in the certificate of incorporation. |
Under the CBCA, dividends may be declared on the common shares at the discretion of the board of directors. Any dividends declared shall be subject to the rights, if any, of shareholders holding shares with special rights as to dividends.
Dividends may not be declared if there are reasonable grounds for believing that the corporation is, or would after the payment be, unable to pay its liabilities as they become due or the realizable value of the corporation’s assets would thereby be less than the aggregate of its liabilities and stated capital of all classes. |
|
|
Number and Election of Directors |
||
| Delaware | Canada | |
|
Under the DGCL, the board of directors must consist of at least one person, and the number of directors is generally fixed by, or in the manner provided in, the bylaws of the corporation, unless the certificate of incorporation fixes the number of directors, in which case a change in the number of directors shall be made only by amendment of the certificate.
The Board may be divided into three classes of directors, with one-third of each class subject to election by the stockholder each year after such classification becomes effective. |
Pursuant to the CBCA, a distributing corporation, any of the issued securities of which remain outstanding and are held by more than one person, shall have no fewer than three directors, at least two of whom are not officers or employees of the corporation or its affiliates. The articles of incorporation will commonly set out the number of initial directors and, if applicable, the minimum and maximum number of directors of the corporation. The shareholders may amend the articles to increase or decrease the number of directors or the minimum or maximum number of directors.
Shareholders may elect directors to hold office for a term expiring not later than the third annual meeting of the shareholders following the election. |
|
Removal of Directors |
||
| Delaware | Canada | |
|
Under the DGCL, any or all directors may be removed with or without cause by the holders of a majority of shares entitled to vote at an election of directors unless the certificate of incorporation otherwise provides or in certain other circumstances if the corporation has cumulative voting. |
Under the CBCA, the shareholders of a corporation may by ordinary resolution remove any director or directors from office. If the holders of any class or series of shares of a corporation have an exclusive right to elect one or more directors, a director so elected may only be removed by shareholders of that class or series. |
|
|
Vacancies on the Board of Directors |
||
| Delaware | Canada | |
|
Under the DGCL, vacancies and newly created directorships resulting from an increase in the authorized number of directors, may be filled by a majority of the directors then in office, although less than a quorum, or by a sole remaining director. |
Under the CBCA, vacancies on the board may be filled by a quorum of directors, except a vacancy resulting from an increase in the number or the minimum or maximum number of directors or a failure to elect the number or minimum number of directors provided for in the articles.
If there is not a quorum of directors or if there has been a failure to elect the number or minimum number of directors provided for in the articles, the directors then in office shall without delay call a special meeting of shareholders to fill the vacancy and, if they fail to call a meeting or if there are no directors then in office, the meeting may be called by any shareholder. |
|
|
Qualifications of Directors |
||
|
Delaware |
Canada |
|
|
Under the DGCA, directors are required to be natural persons, but are not required to be residents of Delaware. The certificate of incorporation or bylaws may prescribe other qualifications for directors. |
Under the CBCA, at least 25% of directors of a CBCA corporation must be resident Canadians. The articles of incorporation may prescribe other qualifications for directors. |
|
|
Board of Director Quorum and Vote Requirements |
||
| Delaware | Canada | |
|
Under the DGCL, a majority of the total number of directors shall constitute a quorum for the transaction of business unless the certificate or bylaws require a greater number. The bylaws may lower the number required for a quorum to one-third the number of directors, but no less.
Under the DGCL, the board of directors may take action by the majority vote of the directors present at a meeting at which a quorum is present unless the certificate of incorporation or bylaws require a greater vote. |
Under the CBCA, a majority of the number of directors or minimum number of directors required by the articles constitutes a quorum at any meeting.
Under the CBCA, directors may not transact business at a meeting of directors unless at least 25% of the directors present are resident Canadians or, if the corporation has less than 4 directors, at least one of the directors present is a resident Canadian, or, if a resident Canadian director who is unable to be present approves in writing, or by telephonic, electronic or other communication facility, the business transacted at the meeting, and the required number of resident Canadian directors would have been present had that director been present at the meeting. |
|
Transactions with Directors and Officers |
||
| Delaware | Canada | |
|
The DGCL generally provides that no transaction between a corporation and one or more of its directors or officers, or between a corporation and any other corporation or other organization in which one or more of its directors or officers, are directors or officers, or have a financial interest, shall be void or voidable solely for this reason, or solely because the director or officer is present at or participates in the meeting of the board or committee which authorizes the transaction, or solely because any such director’s or officer’s votes are counted for such purpose, if (i) the material facts as to the director’s or officer’s interest and as to the transaction are known to the board of directors or the committee, and the board or committee in good faith authorizes the transaction by the affirmative votes of a majority of the disinterested directors, even though the disinterested directors be less than a quorum (ii) the material facts as to the director’s or officer’s interest and as to the transaction are disclosed or are known to the stockholders entitled to vote thereon, and the transaction is specifically approved in good faith by vote of the stockholders; or (iii) the transaction is fair as to the corporation as of the time it is authorized, approved or ratified, by the board of directors, a committee or the stockholders. |
Under the CBCA, a director who holds a disclosable interest in a material contract or transaction into which a corporation has entered or proposes to enter may generally not vote on any directors’ resolution to approve the contract or transaction. A director or officer has a disclosable interest in a material contract or transaction if the director or officer (a) is a party to the contract or transaction; (b) is a director or officer, or an individual acting in a similar capacity, of a party to the contract or transaction; or (c) has a material interest in a party to the contract or transaction.
Under the CBCA, directors do not have to abstain from voting on matters related to director compensation. |
|
|
Limitation on Liability of Directors |
||
| Delaware | Canada | |
| The DGCL permits a corporation to include a provision in its certificate of incorporation eliminating or limiting the personal liability of a director to the corporation or its stockholders for monetary damages for a breach of the director’s fiduciary duty as a director, except for liability. | No provision in a contract, the articles, the by-laws or a resolution may relieve a director or officer from the duty to act in accordance with the CBCA or the regulations or relieves them from liability for a breach thereof. |
|
● |
for breach of the director’s duty of loyalty to the corporation or its stockholders; |
|
● |
for acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of the law; |
|
● |
under Section 174 of the DGCL, which concerns unlawful payment of dividends, stock purchases or redemptions; or |
|
● |
for any transaction from which the director derived an improper personal benefit. |
|
Indemnification of Directors and Officers |
||
| Delaware | Canada | |
|
Under the DGCL, a corporation may indemnify any person who is made a party to any third-party action, suit or proceeding on account of being a director, officer, employee or agent of the corporation (or was serving at the request of the corporation in such capacity for another corporation, partnership, joint venture, trust or other enterprise) against expenses, including attorney's fees, judgments, fines and amounts paid in settlement actually and reasonably incurred by him or her in connection with the action, suit or proceeding through, among other things, a majority vote of a quorum consisting of directors who were not parties to the suit or proceeding, if the person: |
Under the CBCA, a corporation may indemnify a director or officer of the corporation, a former director or officer of the corporation or another individual who acts or acted at the corporation’s request as a director or officer, or an individual acting in a similar capacity, of another entity, against all costs, charges and expenses, including an amount paid to settle an action or satisfy a judgment, reasonably incurred by the individual in respect of any civil, criminal, administrative, investigative or other proceeding in which the individual is involved because of that association with the corporation or other entity. A corporation may not indemnity an individual unless the individual: |
|
●
● ● |
acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the best interests of the corporation; or, in some circumstances, at least not opposed to its best interests; and in a criminal proceeding, had no reasonable cause to believe his or her conduct was unlawful. |
●
● |
acted honestly and in good faith with a view to the best interests of the corporation ; and in the case of a criminal or administrative action or proceeding that is enforced by a monetary penalty, the individual had reasonable grounds for believing that the individual's conduct was lawful. |
| The DGCL permits indemnification for derivative suits against expenses (including legal fees) if the person acted in good faith and in a manner the person reasonably believed to be in or not opposed to the best interests of the corporation, and only if the person is not found liable, unless a court determines the person is fairly and reasonably entitled to the indemnification. | The CBCA permits indemnification for derivative suits with the approval of the court, or if the individual was not judged by the court or other competent authority to have committed any fault or omitted to do anything that the individual ought to have done, acted honestly and in good faith with a view to the best interests of the corporation; and, in the case of a criminal or administrative action or proceeding that is enforced by a monetary penalty, the individual had reasonable grounds for believing that the individual's conduct was lawful. |
|
Call and Notice of Stockholder Meetings |
||
| Delaware | Canada | |
|
Under the DGCL, an annual or special stockholder meeting is held on such date, at such time and at such place as may be designated by the board of directors or any other person authorized to call such meeting under the corporation’s certificate of incorporation or bylaws.
If an annual meeting for election of directors is not held on the date designated or an action by written consent to elect directors in lieu of an annual meeting has not been taken within 30 days after the date designated for the annual meeting, or if no date has been designated, for a period of 13 months after the later of the last annual meeting or the last action by written consent to elect directors in lieu of an annual meeting, the Delaware Court of Chancery may summarily order a meeting to be held upon the application of any stockholder or director.
Special meetings of the stockholders may be called by the board of directors or by such person or persons as may be authorized by the certificate of incorporation or by the bylaws. |
Under the CBCA, the directors are required to call an annual meeting of shareholders not later than 18 months after the corporation comes into existence, and subsequently, not later than 15 months after holding the last preceding annual meeting (but no later than 6 months after the end of the corporation’s preceding financial year). The CBCA requires that a meeting of shareholders may be held anywhere in Canada as the bylaws or board of directors may determine. A meeting of shareholders may be held at a place outside Canada if the place is specified in the articles or all the shareholders entitled to vote at the meeting agree that the meeting is to be held at that place.
The directors may at any time call a special meeting of the shareholders. The holders of not less than five per cent of the issued shares of a corporation that carry the right to vote at a meeting may requisition the directors to call a meeting of shareholders for the purposes stated in the requisition. |
|
|
Stockholder Action by Written Consent |
||
| Delaware | Canada | |
|
Under the DGCL, a majority of the stockholders of a corporation may act by written consent without a meeting unless such action is prohibited by the corporation’s certificate of incorporation. |
Under the CBCA, shareholders may act by written resolution signed by all the shareholders entitled to vote on that resolution at a meeting of shareholders. |
|
|
Stockholder Nominations and Proposals |
||
| Delaware | Canada | |
|
Under the DGCL, the bylaws of a corporation may include provisions respecting the nomination of directors or proposals by stockholders, including requirements for advance notice to the corporation. |
Under the CBCA, a registered holder or beneficial owner of shares that are entitled to be voted at an annual meeting of shareholders may submit to the corporation notice of any matter that the person proposes to raise at the meeting (a “proposal”).
A proposal may include nominations for the election of directors if the proposal is signed by one or more holders of shares representing in the aggregate not less than five per cent of the shares or five per cent of the shares of a class of shares of the corporation entitled to vote at the meeting to which the proposal is to be presented, but this subsection does not preclude nominations made at a meeting of shareholders. |
|
Stockholder Quorum and Vote Requirements |
||
| Delaware | Canada | |
|
Under the DGCL, quorum for a stock corporation is a majority of the shares entitled to vote at the meeting unless the certificate of incorporation or bylaws specify a different quorum, but in no event may a quorum be less than one-third of the shares entitled to vote. Unless the DGCL, certificate of incorporation or bylaws provide for a greater vote, generally the required vote under the DGCL is a majority of the shares present in person or represented by proxy, except for the election of directors which requires a plurality of the votes cast. |
Unless the by-laws otherwise provide, under the CBCA a quorum of shareholders is present at a meeting of shareholders, irrespective of the number of persons actually present at the meeting, if the holders of a majority of the shares entitled to vote at the meeting are present in person or represented by proxy. Under our by-laws, the presence at a shareholder meeting, in person or represented by proxy, of at least two shareholders holding not less than 33 1/3% of the outstanding voting common shares shall constitute a quorum for the purpose of transacting business at the shareholder meeting.
Unless the CBCA, articles of incorporation or bylaws provide for a greater vote, generally the required vote under the CBCA is by ordinary resolution, or a resolution passed by a majority of the votes cast by the shareholders who voted in respect of that resolution. |
|
|
Amendment of Governing Instrument |
||
| Delaware | Canada | |
|
Amendment of Certificate of Incorporation. Generally, under the DGCL, the affirmative vote of the holders of a majority of the outstanding stock entitled to vote is required to approve a proposed amendment to the certificate of incorporation, following the adoption of the amendment by the board of directors of the corporation, provided that the certificate of incorporation may provide for a greater vote. Under the DGCL, holders of outstanding shares of a class or series are entitled to vote separately on an amendment to the certificate of incorporation if the amendment would have certain consequences, including changes that adversely affect the rights and preferences of such class or series.
Amendment of Bylaws. Under the DGCL, after a corporation has received any payment for any of its stock, the power to adopt, amend or repeal bylaws shall be vested in the stockholders entitled to vote; provided, however, that any corporation may, in its certificate of incorporation, provide that bylaws may be adopted, amended or repealed by the board of directors. The fact that such power has been conferred upon the board of directors shall not divest the stockholders of the power nor limit their power to adopt, amend or repeal the bylaws. |
Amendment to Articles of Incorporation. Under the CBCA, either a director or a shareholder entitled to vote at an annual meeting of shareholders may make a proposal to amend the articles. A proposed amendment to the articles requires approval by special resolution of the shareholders. A special resolution is a resolution passed by a majority of not less than two-thirds of the votes cast by the shareholders who voted in respect of the resolution or signed by all shareholders entitled to vote on that resolution.
Under the CBCA, the holders of outstanding shares of a class or series are entitled to vote separately on an amendment to the articles of incorporation if the articles would have certain consequences, including increasing or decreasing the number of shares of such class, or changes that affect the rights and preferences of such class or series.
Amendment of Bylaws. Under the CBCA, a shareholder entitled to vote at an annual meeting of shareholders may make a proposal to make, amend or repeal a by-law. Unless the articles, by-laws or a unanimous shareholder agreement otherwise provide, the directors may, by resolution, make, amend or repeal any by-laws that regulate the business or affairs of the corporation. The directors shall then submit such by-law, or amendment or repeal of a by-law, to the shareholders at the next meeting of shareholders, and the shareholders may, confirm, reject or amend the by-law, amendment or repeal by ordinary resolution. |
|
Votes on Mergers, Consolidations and Sales of Assets |
||
| Delaware | Canada | |
|
The DGCL provides that, unless otherwise provided in the certificate of incorporation or bylaws, the adoption of a merger agreement requires the approval of a majority of the outstanding stock of the corporation entitled to vote thereon. |
Under the CBCA, the approval of an amalgamation agreement requires approval by special resolution. |
|
|
Dissenter’s Rights of Appraisal |
||
| Delaware | Canada | |
|
Under the DGCL, a stockholder of a Delaware corporation generally has the right to dissent from and request payment for the stockholders shares upon a merger or consolidation in which the Delaware corporation is participating, subject to specified procedural requirements, including that such dissenting stockholder does not vote in favor of the merger or consolidation. However, the DGCL does not confer appraisal rights, in certain circumstances, including if the dissenting stockholder owns shares traded on a national securities exchange and will receive publicly traded shares in the merger or consolidation. Under the DGCL, a stockholder asserting appraisal rights does not receive any payment for his or her shares until the court determines the fair value or the parties otherwise agree to a value. The costs of the proceeding may be determined by the court and assessed against the parties as the court deems equitable under the circumstances. |
Under the CBCA, a shareholder may dissent from a transaction and obtain a right of appraisal when the corporation resolves to: (a) amend its articles to add, change or remove any provisions restricting or constraining the issue, transfer or ownership of shares of that class; (b) amend its articles to add, change or remove any restriction on the business or businesses that the corporation may carry on; (c) amalgamate with another entity (other than a short form merger); (d) be continued under the laws of another jurisdiction; (e) sell, lease or exchange all or substantially all its property or assets; or (f) carry out a going-private transaction or a squeezeout transaction. Further, the holders of a class or series of shares entitled to vote as a separate class on an amendment to the articles of incorporation may dissent from such amendment, and this right to dissent applies even if there is only one class of shares.
A shareholder asserting dissenters rights is entitled, subject to specified procedural requirements, including objecting to the action giving rise to dissenters rights and making a proper demand for payment, to be paid by the corporation the fair value of the shares in respect of which the shareholder dissents, determined as of the close of business on the day before the resolution was adopted or the order was made. Under the CBCA, if the shareholder and the corporation do not agree on the fair value for the shareholders shares, the corporation or the dissenting shareholder may apply to a court to fix a fair value for the shares. The court may in its discretion allow a reasonable rate of interest on the amount payable to each dissenting shareholder from the date the action approved by the resolution is effective until the date of payment. |
|
|
Anti-Takeover and Ownership Provisions |
||
| Delaware | Canada | |
|
Unless an issuer opts out of the provisions of Section 203 of the DGCL, Section 203 generally prohibits a public Delaware corporation from engaging in a “business combination” with a holder of 15% or more of the corporation’s voting stock (as defined in Section 203), referred to as an interested stockholder, for a period of three years after the date of the transaction in which the interested stockholder became an interested stockholder, except as otherwise provided in Section 203. For these purposes, the term “business combination” includes mergers, assets sales and other similar transactions with an interested stockholder. |
The CBCA contains no restriction on adoption of a shareholder rights plan. The CBCA does not restrict related party transactions; however, in Canada takeovers and other related party transactions are addressed in provincial securities legislation and policies. |
SHARES ELIGIBLE FOR FUTURE SALE
Future sales of substantial amounts of our common shares in the public market, or the anticipation of such sales, could adversely affect prevailing market prices of our common shares from time to time and could impair our future ability to raise equity capital in the future. Furthermore, because only a limited number of common shares will be available for sale shortly after this offering due to certain contractual and legal restrictions on resale described below, sales of substantial amounts of our common shares in the public market after such restrictions lapse, or the anticipation of such sales, could adversely affect the prevailing market price of our common shares and our ability to raise equity capital in the future.
Upon completion of this offering, we will have outstanding a total of common shares (or shares if the underwriter’s option to purchase additional shares is exercised in full), based on our outstanding common shares as of September 30, 2018, assuming the issuance of common shares in this offering (or shares if the underwriter’s option to purchase additional shares is exercised in full). All of the shares sold in this offering (plus any shares sold as a result of the underwriters’ exercise of their option) will be freely tradable without restriction or further registration under the Securities Act, unless those shares are purchased by our affiliates as that term is defined in Rule 144 under the Securities Act.
Upon completion of this offering, up to 7,856,876 common shares outstanding after this offering will be “restricted securities” under Rule 144. Of these restricted securities, 2,550,736 common shares will be subject to transfer restrictions for 180 days from the date of this prospectus pursuant to lock-up agreements. Restricted securities may be sold in the public market only if they have been registered or if they qualify for an exemption from registration under Rules 144 or 701 or otherwise under the Securities Act.
As of November 15, 2018, we had outstanding options to purchase an aggregate of 639,359 common shares, warrants to purchase an aggregate of 807,563 common shares and deferred share units that will be converted into an aggregate of 21,183 common shares.
We may issue common shares from time to time as consideration for future acquisitions, investments or other corporate purposes. In the event that any such acquisition, investment or other transaction is significant, the number of common shares that we may issue may in turn be significant. We may also grant registration rights covering those common shares issued in connection with any such acquisition and investment.
Rule 144
In general, under Rule 144 of the Securities Act, as in effect on the date of this prospectus, beginning 90 days after the date of this prospectus any person who is not our affiliate at any time during the preceding three months, and who has beneficially owned their shares for at least six months, including the holding period of any prior owner other than one of our affiliates, would be entitled to sell an unlimited number of our common shares provided current public information about us is available, and, after owning such shares for at least one year, including the holding period of any prior owner other than one of our affiliates, would be entitled to sell an unlimited number of our common shares without restriction.
Beginning 90 days after the date of this prospectus, a person who is our affiliate or who was our affiliate at any time during the preceding three months, and who has beneficially owned restricted securities for at least six months, including the holding period of any prior owner other than one of our affiliates, is entitled to sell within any three-month period a number of shares that does not exceed the greater of:
|
● |
1% of the number of our common shares then outstanding, which will equal approximately shares, based on the number of our common shares outstanding upon completion of this offering (or shares if the underwriter exercises its over-allotment option in full); or |
|
● |
the average weekly trading volume of our common stock on The Nasdaq Capital Market during the four calendar weeks preceding the filing of a Notice of Proposed Sale of Securities pursuant to Rule 144 with respect to the sale. |
Sales under Rule 144 by our affiliates are also subject to manner of sale provisions and notice requirements and to the availability of current public information about us.
Upon expiration of the 180-day lock-up period described below, common shares will be eligible for sale under Rule 144, assuming an initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus. We cannot estimate the number of common shares that our existing shareholders will elect to sell under Rule 144.
Rule 701
Rule 701 generally allows a shareholder who purchased our common shares pursuant to a written compensatory plan or contract and who is not deemed to have been an affiliate of our company during the immediately preceding 90 days to sell these shares in reliance upon Rule 144, but without being required to comply with the public information, holding period, volume limitation or notice provisions of Rule 144. Rule 701 also permits affiliates of our company to sell their Rule 701 shares under Rule 144 without complying with the holding period requirements of Rule 144. All holders of Rule 701 shares, however, are required by that rule to wait until 90 days after the date of this prospectus before selling those shares pursuant to Rule 701.
Lock-Up Agreements
We expect that our officers, directors, and certain shareholders will enter into an agreement that, without the prior written consent of the underwriter, they will not, subject to limited exceptions, directly or indirectly sell or dispose of any of our common shares or any securities convertible into or exchangeable or exercisable for our common shares for a period of 180 days after the date of this prospectus. The lock-up restrictions and specified exceptions are described in more detail under “Underwriting.”
Form S-8 Registration Statement
As soon as practicable after the closing of this offering, we intend to file one or more registration statements on Form S-8 under the Securities Act covering all of the common shares subject to outstanding options and deferred share units and the common shares reserved for issuance under our current stock option plan. We expect to file these registration statements, as applicable, as soon as permitted under the Securities Act. However, the shares registered on Form S-8 may be subject to the volume limitations and the manner of sale, notice and public information requirements of Rule 144 and will not be eligible for resale until expiration of the lock-up and market standoff agreements to which they are subject. Of the 639,359 common shares that were subject to stock options outstanding as of September 30, 2018, options to purchase 346,480 common shares were vested as of September 30, 2018. Of the stock options outstanding as of September 30, 2018, options to purchase 495,725 common shares will not be eligible for sale until expiration of the 180 day lock-up and market standoff agreements to which they are subject.
Registration Rights
We have not granted any rights to register our common shares under the Securities Act.
CERTAIN UNITED STATES FEDERAL INCOME TAX CONSIDERATIONS
The following discussion is generally limited to certain material U.S. federal income tax considerations relating to the purchase, ownership and disposition of the common shares by U.S. Holders (as defined below). This discussion applies to U.S. Holders that hold common shares as capital assets. This summary is for general information purposes only and does not purport to be a complete analysis or listing of all potential U.S. federal income tax considerations that may apply to a U.S. Holder arising from and relating to the acquisition, ownership, and disposition of common shares. Accordingly, this summary is not intended to be, and should not be construed as, legal or U.S. federal income tax advice with respect to any U.S. Holder. Although this discussion is generally limited to the U.S. federal income tax considerations to U.S. Holders the U.S. federal income tax treatment of dividends on and gain on sale or exchange of our common shares by certain “Non-U.S. Holders” (as defined below) is included below at “U.S. Federal Income Taxation of Non-U.S. Holders.”
No legal opinion from U.S. legal counsel or ruling from the Internal Revenue Service (the “IRS”) has been requested, or will be obtained, regarding the U.S. federal income tax consequences of the acquisition, ownership, and disposition of common shares. This summary is not binding on the IRS, and the IRS is not precluded from taking a position that is different from, and contrary to, the positions presented in this summary. In addition, because the guidance on which this summary is based are subject to various interpretations, the IRS and the U.S. courts could disagree with one or more of the positions described in this summary.
This discussion is based on the U.S. Internal Revenue Code of 1986, as amended (the “Code”), U.S. Treasury regulations promulgated thereunder and administrative and judicial interpretations thereof, all as in effect on the date hereof and all of which are subject to change, possibly with retroactive effect. This summary does not discuss the potential effects, whether adverse or beneficial, of any proposed legislation.
This discussion does not address all of the U.S. federal income tax considerations that may be relevant to specific U.S. Holders in light of their particular circumstances or to U.S. Holders subject to special treatment under U.S. federal income tax law (such as certain financial institutions, insurance companies, broker-dealers and traders in securities or other persons that generally mark their securities to market for U.S. federal income tax purposes, tax-exempt entities, retirement plans, regulated investment companies, real estate investment trusts, certain former citizens or residents of the United States, persons who hold common shares as part of a “straddle,” “hedge,” “conversion transaction,” “synthetic security” or integrated investment, persons that have a “functional currency” other than the U.S. dollar, persons that own (or are deemed to own) 10% or more (by voting power or value) of our common shares, corporations that accumulate earnings to avoid U.S. federal income tax, partnerships and other pass-through entities, and investors in such pass-through entities). This discussion does not address any U.S. state or local or non-U.S. tax considerations or any U.S. federal estate, gift or alternative minimum tax considerations. In addition, except as specifically set forth below, this summary does not discuss applicable tax reporting requirements.
As used in this discussion, the term “U.S. Holder” means a beneficial owner of common shares that is, for U.S. federal income tax purposes, (1) an individual who is a citizen or resident of the United States, (2) a corporation (or entity treated as a corporation for U.S. federal income tax purposes) created or organized in or under the laws of the United States, any state thereof, or the District of Columbia, (3) an estate the income of which is subject to U.S. federal income tax regardless of its source or (4) a trust (x) with respect to which a court within the United States is able to exercise primary supervision over its administration and one or more United States persons have the authority to control all of its substantial decisions or (y) that has elected under applicable U.S. Treasury regulations to be treated as a domestic trust for U.S. federal income tax purposes.
If an entity treated as a partnership for U.S. federal income tax purposes holds the common shares, the U.S. federal income tax considerations relating to an investment in the common shares will depend in part upon the status and activities of such entity and the particular partner. Any such entity should consult its own tax advisor regarding the U.S. federal income tax considerations applicable to it and its partners of the purchase, ownership and disposition of the common shares.
Persons holding common shares should consult their own tax advisors as to the particular tax considerations applicable to them relating to the purchase, ownership and disposition of common shares, including the applicability of U.S. federal, state and local tax laws and non-U.S. tax laws.
Distributions
Subject to the discussion below under “Passive Foreign Investment Company Considerations,” a U.S. Holder that receives a distribution with respect to the common shares generally will be required to include the gross amount of such distribution (before reduction for any Canadian withholding taxes) in gross income as a dividend when actually or constructively received to the extent of the U.S. Holder’s pro rata share of our current and/or accumulated earnings and profits (as determined under U.S. federal income tax principles). To the extent a distribution received by a U.S. Holder is not a dividend because it exceeds the U.S. Holder’s pro rata share of our current and accumulated earnings and profits, it will be treated first as a tax-free return of capital and reduce (but not below zero) the adjusted tax basis of the U.S. Holder’s common shares. To the extent the distribution exceeds the adjusted tax basis of the U.S. Holder’s common shares, the remainder will be taxed as capital gain. However, we cannot provide any assurance that we will maintain or provide earnings and profits determinations in accordance with U.S. federal income tax principles. Therefore, U.S. Holders should expect that a distribution will generally be treated as a dividend even if that distribution would otherwise be treated as a non-taxable return of capital or as capital gain under the rules described above.
The U.S. dollar value of any distribution on the common shares made in Canadian dollars generally should be calculated by reference to the exchange rate between the U.S. dollar and the Canadian dollar in effect on the date of receipt (or deemed receipt) of such distribution by the U.S. Holder regardless of whether the Canadian dollars so received are in fact converted into U.S. dollars at that time. If the Canadian dollars received are converted into U.S. dollars on the date of receipt (or deemed receipt), a U.S. Holder generally should not recognize currency gain or loss on such conversion. If the Canadian dollars received are not converted into U.S. dollars on the date of receipt (or deemed receipt), a U.S. Holder generally will have a basis in such Canadian dollars equal to the U.S. dollar value of such Canadian dollars on the date of receipt (or deemed receipt). Any gain or loss on a subsequent conversion or other disposition of such Canadian dollars by such U.S. Holder generally will be treated as ordinary income or loss and generally will be income or loss from sources within the United States for U.S. foreign tax credit purposes. Different rules apply to U.S. Holders who use the accrual method of tax accounting. Each U.S. Holder should consult its own U.S. tax advisors regarding the U.S. federal income tax consequences of receiving, owning, and disposing of foreign currency.
Distributions on the common shares that are treated as dividends generally will constitute income from sources outside the United States for foreign tax credit purposes and generally will constitute “passive category income.” Because we are not a United States corporation, such dividends will not be eligible for the “dividends received” deduction generally allowed to corporate shareholders with respect to dividends received from U.S. corporations. Dividends paid by a “qualified foreign corporation” to a U.S. Holder who is an individual, trust or estate will generally be treated as “qualified dividend income” and are eligible for taxation at a reduced capital gains rate rather than the marginal tax rates generally applicable to ordinary income provided that a holding period requirement (more than 60 days of ownership, without protection from the risk of loss, during the 121-day period beginning 60 days before the ex-dividend date) and certain other requirements are met. However, if we are a passive foreign investment company (“PFIC”) for the taxable year in which the dividend is paid or the preceding taxable year (see discussion below under “Passive Foreign Investment Company Considerations”), we will not be treated as a qualified foreign corporation, and therefore the reduced capital gains tax rate described above will not apply. Each U.S. Holder is advised to consult its own tax advisors regarding the availability of the reduced tax rate on dividends.
If a U.S. Holder is subject to Canadian withholding tax on dividends paid on the holder’s common shares (see discussion below under “Certain Canadian Federal Income Tax Considerations—Dividends”), the U.S. Holder may be eligible, subject to a number of complex limitations, to claim a credit against its U.S. federal income tax for the Canadian withholding tax imposed on the dividends. A U.S. Holder may claim a deduction for the Canadian withholding tax in lieu of a credit, but only for a year in which the U.S. Holder elects to do so for all creditable foreign income taxes. The rules governing the foreign tax credit are complex. Each U.S. Holder is advised to consult its tax advisor regarding the availability of the foreign tax credit under its particular circumstances.
Sale, Exchange or Other Disposition of Common Shares
Subject to the discussion below under “Passive Foreign Investment Company Considerations,” a U.S. Holder generally will recognize capital gain or loss for U.S. federal income tax purposes upon the sale, exchange or other disposition of common shares. The amount of gain recognized will equal the excess of the amount realized (i.e., the amount of cash plus the fair market value of any property received) over the U.S. Holder’s adjusted tax basis in the common shares sold or exchanged. The amount of loss recognized will equal the excess of the U.S. Holder’s adjusted tax basis in the common shares sold or exchanged over the amount realized. Such capital gain or loss generally will be long-term capital gain or loss if, on the date of sale, exchange or other disposition, the common shares were held by the U.S. Holder for more than one year. Net long-term capital gain derived by a non-corporate U.S. Holder currently is subject to tax at reduced rates. The deductibility of a capital loss is subject to limitations. Any gain or loss recognized from the sale, exchange or other disposition of common shares will generally be gain or loss from sources within the United States for U.S. foreign tax credit purposes, except as otherwise provided in an applicable income tax treaty and if an election is properly made under the Code.
Passive Foreign Investment Company Considerations
In general, a corporation organized outside the United States will be treated as a PFIC in any taxable year in which either (1) at least 75% of its gross income is “passive income” or (2) at least 50% of the average quarterly value of its assets is attributable to assets that produce passive income or are held for the production of passive income. Passive income for this purpose generally includes, among other things, dividends, interest, royalties, rents, and gains from commodities transactions and from the sale or exchange of property that gives rise to passive income. Assets that produce or are held for the production of passive income include cash, even if held as working capital or raised in a public offering, marketable securities and other assets that may produce passive income. The average percentage of a corporation’s assets that produce or are held for the production of passive income generally is determined on the basis of the fair market value of the corporation’s assets at the end of each quarter (which may be determined in part by the market value of our common shares, which is subject to change). In determining whether a foreign corporation is a PFIC, a proportionate share of the items of gross income and assets of each corporation in which it owns, directly or indirectly, at least a 25% interest (by value) are taken into account.
Based on the price of our common shares and the composition of our gross assets (i) we believe that we were a PFIC for the taxable year ended December 31, 2016, (ii) we do not believe that we were a PFIC for the taxable year ended December 31, 2017 and (iii) we do not believe that we will be a PFIC for the taxable year ending December 31, 2018. Our status as a PFIC is a fact-intensive determination made on an annual basis, and we cannot provide any assurance regarding our PFIC status for the taxable year ending December 31, 2018 or for future taxable years. No opinion of legal counsel or ruling from the IRS concerning our status as a PFIC has been obtained or is currently planned to be requested. However, the determination of our PFIC status is made annually after the close of each taxable year and it is difficult to predict before such determination whether we will be a PFIC for any given taxable year. Even if we determine that we are not a PFIC after the close of a taxable year, there can be no assurance that the IRS will agree with our conclusion. No assurance can be provided regarded our PFIC status, and neither we nor our United States counsel expresses any opinion with respect to our PFIC status.
If we are a PFIC at any time when a non-corporate U.S. Holder owns common shares, such U.S. Holder will generally be subject to federal tax under the excess distribution rules (described below). Under such rules, additional taxes and interest charges would apply to certain distributions by us or to gain upon dispositions of our common shares if such U.S. Holder has not elected to have his or her investment in our common shares treated as an investment in a “qualified electing fund” or has not made a “mark-to-market election.” If neither of such elections are made, the excess distribution rules apply to (1) distributions paid during a taxable year that are greater than 125% of the average annual distributions paid in the three preceding taxable years, or, if shorter, the U.S. Holder’s holding period for the common shares, and (2) any gain recognized on a sale, exchange or other disposition (which would include a pledge) of common shares. Under the excess distribution rules, the non-corporate U.S. Holder’s tax liability will be determined by allocating such distribution or gain ratably to each day in the U.S. Holder’s holding period for the common shares. The amount allocated to the current taxable year (i.e., the year in which the distribution occurs or the gain is recognized) and any year prior to the first taxable year in which we were a PFIC in the holding period will be taxed as ordinary income earned in the current taxable year. The amount allocated to other taxable years (i.e., prior years in which we were a PFIC) will be taxed at the highest marginal rate in effect (for individuals or corporations as applicable) for ordinary income in each such taxable year, and an interest charge, generally that applicable to the underpayment of tax, will be added to the tax. These adverse tax consequences would not apply to a pension or profit sharing trust or other tax-exempt organization that did not borrow funds or otherwise utilize leverage in connection with its acquisition of our common shares. In addition, if a Non-Electing Holder who is an individual dies while owning our common such, such holder's successor generally would not receive a step-up in tax basis with respect to such common shares.
If we are a PFIC at any time when a U.S. Holder holds our common shares, we will generally continue to be treated as a PFIC with respect to the U.S. Holder for all succeeding years during which the U.S. Holder holds our common shares even if we cease to meet the PFIC gross income test or asset test. However, if we cease to meet these tests, a U.S. Holder can avoid the continuing impact of the PFIC rules by making a special election (a “Purging Election”) to recognize gain by making a “deemed sale” election with respect to all of the U.S. Holder’s common shares and have such common shares deemed to be sold at their fair market value on the last day of the last taxable year during which we were a PFIC. In addition, for a U.S. Holder making such an election, a new holding period would be deemed to begin for our common shares for purposes of the PFIC rules. After the Purging Election, the common shares with respect to which the Purging Election was made will not be treated as shares in a PFIC unless we subsequently become a PFIC.
The tax considerations that would apply if we were a PFIC would be different from those described above if a U.S. Holder were able to make a valid “qualified electing fund,” or “QEF election.” For each year that we meet the PFIC gross income test or asset test, an electing U.S. Holder would be required to include in gross income, its pro rata share of our ordinary income and net capital gains, if any, as determined under U.S. federal income tax principles. The U.S. Holder’s adjusted tax basis in our common shares would be increased by the amount of such inclusions. An actual distribution to the U.S. Holder out of such income generally would not be treated as a dividend and would decrease the U.S. Holder’s adjusted tax basis in our common shares. Gain realized from the sale of our common shares covered by a QEF election would be taxed as a capital gain. Generally, a QEF election must be made by the U.S. Holder in a timely filed tax return for the first taxable year in which the U.S. Holder held our common shares that includes the close of our taxable year for which we met the PFIC gross income test or asset test. A QEF election is made on IRS Form 8621. U.S. Holders will be eligible to make QEF elections only if we agree to provide U.S. Holders with the information they will need to comply with the QEF rules. In the event we become a PFIC, we intend to provide all information and documentation that a U.S. Holder making a QEF election is required to obtain for U.S. federal income tax purposes (e.g., the U.S. Holder’s pro rata share of ordinary income and net capital gain, and a “PFIC Annual Information Statement” as described in applicable U.S. Treasury regulations).
A U.S. Holder may also mitigate the adverse tax consequences by timely making a mark-to-market election, provided the U.S. Holder completes and files IRS Form 8621 in accordance with the relevant instructions and related Treasury regulations. A U.S. Holder who makes the mark-to-market election generally must include as ordinary income each year the increase in the fair market value of the common shares and deduct from gross income the decrease in the value of such shares during each of its taxable years, but limited to the amount of previously recognized net gains. The U.S. Holder’s tax basis in the common shares would be adjusted to reflect any income or loss recognized as a result of the mark-to-market election. Any gain from a sale, exchange or other disposition of the common shares in any taxable year in which we are a PFIC (i.e., when we meet the gross income test or asset test described above) would be treated as ordinary income and any loss from a sale, exchange or other disposition would be treated first as an ordinary loss (to the extent of any net mark-to-market gains previously included in income) and thereafter as a capital loss. If we cease to be a PFIC, any gain or loss recognized by a U.S. Holder on the sale or exchange of the common shares would be classified as a capital gain or loss.
A mark-to-market election is available to a U.S. Holder only for “marketable stock.” Generally, stock will be considered marketable stock if it is “regularly traded” on a “qualified exchange” within the meaning of applicable U.S. Treasury regulations. A class of stock is regularly traded during any calendar year during which such class of stock is traded, other than in de minimis quantities, on at least 15 days during each calendar quarter. The common shares should be marketable stock as long as they are listed on The Nasdaq Capital Market or the TSX Venture Exchange and are regularly traded. A mark-to-market election will not apply to the common shares for any taxable year during which we are not a PFIC, but will remain in effect with respect to any subsequent taxable year in which we again become a PFIC. Such election will not apply to any subsidiary that we own. Accordingly, a U.S. Holder may continue to be subject to the PFIC rules with respect to any lower-tier PFICs notwithstanding the U.S. Holder’s mark-to-market election. Whether our common shares are regularly traded on a qualified exchange is an annual determination based on facts that, in part, are beyond our control. Accordingly, a U.S. Holder might not be eligible to make a mark-to-market election to mitigate the adverse tax consequences if we are characterized as a PFIC.
Each U.S. person who is a shareholder of a PFIC generally must file an annual report (on IRS Form 8621) with the IRS containing certain information, and the failure to file such report could result in the imposition of penalties on such U.S. person and in the extension of the statute of limitations with respect to federal income tax returns filed by such U.S. person.
The U.S. federal income tax rules relating to PFICs are very complex. U.S. Holders are urged to consult their own tax advisors with respect to the purchase, ownership and disposition of common shares, the consequences to them of an investment in a PFIC, any elections available with respect to the common shares and the IRS information reporting obligations with respect to the purchase, ownership and disposition of common shares in the event we are considered a PFIC.
Additional Tax on Passive Income
Certain U.S. Holders that are individuals, estates or trusts (other than trusts that are exempt from tax) will be subject to a 3.8% tax on all or a portion of their “net investment income,” which includes dividends on the common shares, and net gains from the disposition of the common shares. Further, excess distributions treated as dividends, gains treated as excess distributions, and mark-to-market inclusions and deductions are all included in the calculation of net investment income.
Treasury regulations provide, subject to the election described in the following paragraph, that solely for purposes of this additional tax, that distributions of previously taxed income will be treated as dividends and included in net investment income subject to the additional 3.8% tax. Additionally, to determine the amount of any capital gain from the sale or other taxable disposition of common shares that will be subject to the additional tax on net investment income, a U.S. Holder who has made a QEF election will be required to recalculate its basis in the common shares excluding QEF election basis adjustments.
Alternatively, a U.S. Holder may make an election which will be effective with respect to all interests in controlled foreign corporations and QEF election held in that year or acquired in future years. Under this election, a U.S. Holder pays the additional 3.8% tax on QEF election income inclusions and on gains calculated after giving effect to related tax basis adjustments. U.S. Holders that are individuals, estates or trusts should consult their own tax advisors regarding the applicability of this tax to any of their income or gains in respect of the common shares.
U.S. Federal Income Taxation of Non-U.S. Holders
A beneficial owner of our common shares, other than a partnership or entity treated as a partnership for U.S. Federal income tax purposes, that is not a U.S. Holder is referred to herein as a “Non-U.S. Holder”. Non-U.S. Holders generally will not be subject to U.S. federal income tax or withholding tax on dividends received from us with respect to our common shares, unless that income is effectively connected with the Non-U.S. Holder's conduct of a trade or business in the United States. In general, if the Non-U.S. Holder is entitled to the benefits of certain U.S. income tax treaties with respect to those dividends, that income is taxable only if it is attributable to a permanent establishment maintained by the Non-U.S. Holder in the United States.
Non-U.S. Holders generally will not be subject to U.S. federal income tax or withholding tax on any gain realized upon the sale, exchange or other disposition of our common shares, unless:
|
● |
the gain is effectively connected with the Non-U.S. Holder's conduct of a trade or business in the United States. In general, if the Non-U.S. Holder is entitled to the benefits of certain income tax treaties with respect to that gain, that gain is taxable only if it is attributable to a permanent establishment maintained by the Non-U.S. Holder in the United States; or |
|
● |
the Non-U.S. Holder is an individual who is present in the United States for 183 days or more during the taxable year of disposition and other conditions are met. |
If the Non-U.S. Holder is engaged in a U.S. trade or business for U.S. federal income tax purposes, the income from the common shares, including dividends and the gain from the sale, exchange or other disposition of the stock, that is effectively connected with the conduct of that trade or business will generally be subject to regular U.S. federal income tax in the same manner as discussed above relating to the general taxation of U.S. Holders. In addition, if you are a corporate Non-U.S. Holder, your earnings and profits that are attributable to the effectively connected income, which are subject to certain adjustments, may be subject to an additional branch profits tax at a rate of 30%, or at a lower rate as may be specified by an applicable U.S. income tax treaty.
Information Reporting with Respect to Foreign Financial Assets
U.S. individuals that own “specified foreign financial assets” (as defined in Section 6038D of the Code) with an aggregate fair market value exceeding certain threshold amounts generally are required to file an information report on IRS Form 8938 with respect to such assets with their tax returns. Significant penalties may apply to persons who fail to comply with these rules. Specified foreign financial assets include not only financial accounts maintained in foreign financial institutions, but also, unless held in accounts maintained by a financial institution, any stock or security issued by a non-U.S. person, such as our common shares. Upon the issuance of future U.S. Treasury regulations, these information reporting requirements may apply to certain U.S. entities that own specified foreign financial assets. The failure to report information required under the current regulations could result in substantial penalties and in the extension of the statute of limitations with respect to federal income tax returns filed by a U.S. Holder. U.S. Holders should consult their own tax advisors regarding the possible implications of these U.S. Treasury regulations for an investment in our common shares.
Special Reporting Requirements for Transfers to Foreign Corporations
A U.S. Holder that acquires common shares generally will be required to file Form 926 with the IRS if (1) immediately after the acquisition such U.S. Holder, directly or indirectly, owns at least 10% of the common shares, or (2) the amount of cash transferred in exchange for common shares during the 12-month period ending on the date of the acquisition exceeds US$100,000. Significant penalties may apply for failing to satisfy these filing requirements. U.S. Holders are urged to contact their tax advisors regarding these filing requirements.
Information Reporting and Backup Withholding
Dividends on and proceeds from the sale or other disposition of common shares may be reported to the IRS unless the U.S. Holder establishes a basis for exemption. Backup withholding may apply to amounts subject to reporting if (1) the U.S. holder fails to provide an accurate taxpayer identification number or otherwise establish a basis for exemption, (2) the U.S. Holder is notified by the IRS that backup withholding applies, or (3) the payment is described in certain other categories of persons.
If you sell your common shares through a U.S. office of a broker, the payment of the proceeds is subject to both U.S. backup withholding and information reporting unless you certify that you are a non-U.S. person, under penalties of perjury, or you otherwise establish an exemption. If you sell your common shares through a non-U.S. office of a non-U.S. broker and the sales proceeds are paid to you outside the United States then information reporting and backup withholding generally will not apply to that payment. However, U.S. information reporting requirements, but not backup withholding, will apply to a payment of sales proceeds, even if that payment is made to you outside the United States, if you sell your common shares through a non-U.S. office of a broker that is a U.S. person or has certain other contacts with the United States, unless you certify that you are a non-U.S. person, under penalty of perjury, or you otherwise establish an exemption.
Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules generally will be allowed as a refund or a credit against a U.S. Holder’s U.S. federal income tax liability if the required information is furnished by the U.S. Holder on a timely basis to the IRS.
THE DISCUSSION ABOVE IS A GENERAL SUMMARY. IT DOES NOT COVER ALL TAX MATTERS THAT MAY BE OF IMPORTANCE TO A U.S. HOLDER. EACH U.S. HOLDER IS URGED TO CONSULT ITS OWN TAX ADVISOR ABOUT THE TAX CONSEQUENCES TO IT OF AN INVESTMENT IN COMMON SHARES IN LIGHT OF THE INVESTOR’S OWN CIRCUMSTANCES.
MATERIAL CANADIAN FEDERAL INCOME TAX CONSIDERATIONS
The following is, as of November 15, 2018, a summary of the principal Canadian federal income tax considerations under the Income Tax Act (Canada) (the “Tax Act ”) generally applicable to a holder of our common shares who, for purposes of the Tax Act and at all relevant times, is neither resident in Canada nor deemed to be resident in Canada for purposes of the Tax Act and any applicable income tax treaty or convention, and who does not use or hold (and is not deemed to use or hold) common shares in the course of carrying on a business in Canada, deals at arm’s length with and is not affiliated with us and holds our common shares as capital property (a “ Holder”). Generally, common shares will be considered to be capital property to a Holder thereof provided that the Holder does not hold common shares in the course of carrying on a business and such Holder has not acquired them in one or more transactions considered to be an adventure or concern in the nature of trade.
This summary does not apply to a Holder (i) that is a “financial institution” for purposes of the mark-to-market rules contained in the Tax Act; (ii) that is a “specified financial institution” as defined in the Tax Act; (iii) an interest in which is a “tax shelter investment” as defined in the Tax Act; or (iv) that has elected to report its tax results in a functional currency other than Canadian currency. Special rules, which are not discussed in this summary, may apply to a Holder that is an “authorized foreign bank” within the meaning of the Tax Act, a partnership or an insurer carrying on business in Canada and elsewhere. Such Holders should consult their own tax advisors.
This summary is based upon the provisions of the Tax Act (including the regulations (“Regulations”) thereunder) in force as of November 6, 2018 and our understanding of the current administrative policies and assessing practices of the Canada Revenue Agency (the “CRA ”) published in writing by the CRA prior to November 6, 2018. This summary takes into account all specific proposals to amend the Tax Act (and the Regulations) publicly announced by or on behalf of the Minister of Finance (Canada) prior to the date hereof (the “Tax Proposals”) and assumes that the Tax Proposals will be enacted in the form proposed, although no assurance can be given that the Tax Proposals will be enacted in their current form or at all. This summary does not otherwise take into account any changes in law or in the administrative policies or assessing practices of the CRA, whether by legislative, governmental or judicial decision or action. This summary is not exhaustive of all possible Canadian federal income tax considerations, and does not take into account other federal or any provincial, territorial or foreign income tax legislation or considerations, which may differ materially from those described in this summary.
This summary is of a general nature only and is not, and is not intended to be, and should not be construed to be, legal or tax advice to any particular Holder, and no representations concerning the tax consequences to any particular Holder are made. Holders should consult their own tax advisors regarding the income tax considerations applicable to them having regard to their particular circumstances.
Dividends
Dividends paid or credited (or deemed to be paid or credited) to a Holder by us are subject to Canadian withholding tax at the rate of 25% unless reduced by the terms of an applicable tax treaty or convention. For example, under the US Treaty, as amended, the dividend withholding tax rate is generally reduced to 15% in respect of a dividend paid or credited to a Holder beneficially entitled to the dividend who is resident in the United States for purposes of the US Treaty and whose entitlement to the benefits of the US Treaty is not limited by the limitation of benefits provisions of the US Treaty. Holders are urged to consult their own tax advisors to determine their entitlement to relief under the US Treaty or any other applicable tax treaty as well as their ability to claim foreign tax credits with respect to any Canadian withholding tax, based on their particular circumstances.
Disposition of Common Shares
A Holder generally will not be subject to tax under the Tax Act in respect of a capital gain realized on the disposition or deemed disposition of a common share, unless the common share constitutes or is deemed to constitute “taxable Canadian property” to the Holder thereof for purposes of the Tax Act, and the gain is not exempt from tax pursuant to the terms of an applicable tax treaty or convention.
In general, provided the common shares are listed on a “designated stock exchange” (which currently includes the TSX Venture Exchange and The Nasdaq Capital Market, if our listing application is accepted by Nasdaq) at the date of the disposition, the common shares will only constitute “taxable Canadian property” of a Holder if, at any time within the 60-month period preceding the disposition: (i) such Holder, persons with whom the Holder did not deal at arm’s length, partnerships in which the Holder or a person with whom the Holder did not deal at arm’s length holds a membership interest directly or indirectly through one or more partnerships, or any combination thereof, owned 25% or more of the issued shares of any class or series of the Company’s share capital; and (ii) more than 50% of the fair market value of the common shares was derived directly or indirectly from one or any combination of (A) real or immovable property situated in Canada, (B) Canadian resource properties, (C) timber resource properties, and (D) options in respect of, or interests in, or for civil law rights in, property described in any of subparagraphs (ii)(A) to (C), whether or not the property exists. However, and despite the foregoing, in certain circumstances the common shares may be deemed to be “taxable Canadian property” under the Tax Act.
Holders whose common shares may be “taxable Canadian property” should consult their own tax advisers.
UNDERWRITING
The underwriter named below has agreed to buy, subject to the terms of the underwriting agreement, the number of common shares listed opposite its name below. The underwriter is committed to purchase and pay for all of the common shares if any are purchased, other than those common shares covered by the over-allotment option described below. Craig-Hallum Capital Group LLC is the sole book-running manager for the offering.
|
Underwriter |
Number of Shares |
|
|
Craig-Hallum Capital Group LLC |
||
|
Total |
The underwriter has advised us that it proposes to offer the common shares to the public at a price of $ per share. The underwriter proposes to offer the common shares to certain dealers at the same price less a concession of not more than $ per share. After the offering, these figures may be changed by the underwriter.
The shares sold in this offering are expected to be ready for delivery against payment in immediately available funds on or about , 2018, subject to customary closing conditions. The underwriter may reject all or part of any order.
We have granted to the underwriter an option to purchase up to an additional common shares from us at the same price to the public, and with the same underwriting discount, as set forth in the table below. The underwriter may exercise this option any time during the 30-day period after the date of this prospectus, but only to cover over-allotments, if any. To the extent the underwriter exercises the option, the underwriter will become obligated, subject to certain conditions, to purchase the shares for which it exercises the option.
Commissions and Discounts
The table below summarizes the underwriting discounts that we will pay to the underwriter. These amounts are shown assuming both no exercise and full exercise of the over-allotment option. In addition to the underwriting discount, we have agreed to pay up to $125,000 of the fees and expenses of the underwriter, which may include the fees and expenses of counsel to the underwriter. In connection with the successful completion of this offering, for the price of $50 the underwriter may purchase a warrant to purchase common shares equal to 5.0% of the common shares sold in this offering at an exercise price that is 120% of the initial public offering price per share in this offering; provided that the underwriter will only receive such warrants relating to the over-allotment option upon the closing (if any) of the over-allotment option. The warrants are exercisable during the period commencing on the date of the prospectus and ending five years from the date of this prospectus and are not exercisable or convertible more than five years from the date of this prospectus as provided in FINRA Rule 5110 (f)(2)(G)(i). The warrants may not be sold during this offering, or sold, transferred, assigned, pledged or hypothecated, or be the subject of any hedging, short sale, derivative, put, or call transaction that would result in the effective economic disposition of the warrants, or the shares acquirable upon exercise thereof, by any person for a period of 180 days immediately following the effective date of the registration statement of which this prospectus form a part, except as provided in paragraph (g)(2) of Rule 5110 of the Financial Industry Regulatory Authority (“FINRA”).
Except as disclosed in this prospectus, the underwriter has not received and will not receive from us any other item of compensation or expense in connection with this offering considered by FINRA to be underwriting compensation under FINRA Rule 5110. The underwriting discount was determined through an arms’ length negotiation between us and the underwriter.
|
Per Share |
Total with Allotment |
Total with Over- Allotment |
||||||||||
|
Underwriting discount to be paid by us |
$ | $ | $ | |||||||||
We estimate that the total expenses of this offering, excluding underwriting discounts, will be approximately $ . This includes $125,000 of fees and expenses of the underwriter. These expenses are payable by us.
Indemnification
We also have agreed to indemnify the underwriter against certain liabilities, including civil liabilities under the Securities Act or to contribute to payments that the underwriter may be required to make in respect of those liabilities.
No Sales of Common Shares
We, each of our directors and officers and certain of our significant shareholders have agreed not to offer, sell, agree to sell, directly or indirectly, or otherwise dispose of any common shares or any securities convertible into or exchangeable for common shares without the prior written consent of the underwriter for a period of 180 days after the date of this prospectus. These lock-up agreements provide limited exceptions and their restrictions may be waived at any time by the underwriter.
Determination of Offering Price
The underwriter has advised us that it proposes to offer the shares directly to the public at the initial public offering price set forth on the cover page of this preliminary prospectus. The initial public offering price is subject to change as a result of market conditions and other factors. Our common shares trade in Canada on the TSX Venture Exchange under the trading symbol “DMA” and over-the-counter in the United States on the OTCQB marketplace under the trading symbol “DMCAF.” The initial public offering price of the shares was determined by negotiation between us and the underwriter. The principal factors considered in determining the initial public offering price of the shares included:
|
● |
historical and recent trading price of our common shares on the TSX Venture Exchange in Canada and on the OTCQB marketplace in the United States; |
|
● |
the information in this prospectus and otherwise available to the underwriter, including our financial information; |
|
● |
the history and the prospects for the industry in which we compete; |
|
● |
the ability and experience of our management; |
|
● |
the prospects for our future earnings; |
|
● |
the present state of our development and our current financial condition; |
|
● |
the general condition of the economy and the securities markets in the United States at the time of this initial public offering; |
|
● |
the recent market prices of, and the demand for, publicly-traded securities of generally comparable companies; and |
|
● |
other factors as were deemed relevant. |
We cannot be sure that the initial public offering price will correspond to the price at which the common shares will trade in the public market following this offering or that an active trading market for the common shares will develop or continue after this offering.
Price Stabilization, Short Positions and Penalty Bids
To facilitate this offering, the underwriter may engage in transactions that stabilize, maintain or otherwise affect the price of our common shares during and after the offering. Specifically, the underwriter may create a short position in our common shares for its own accounts by selling more common shares than we have sold to the underwriter. The underwriter may close out any short position by purchasing common shares in the open market.
In addition, the underwriter may stabilize or maintain the price of our common shares by bidding for or purchasing common shares in the open market and may impose penalty bids. If penalty bids are imposed, selling concessions allowed to broker-dealers participating in this offering are reclaimed if common shares previously distributed in this offering are repurchased, whether in connection with stabilization transactions or otherwise. The effect of these transactions may be to stabilize or maintain the market price of our common shares at a level above that which might otherwise prevail in the open market. The imposition of a penalty bid may also affect the price of our common shares to the extent that it discourages resales of our common shares.
The magnitude or effect of any stabilization or other transactions is uncertain. These transactions may be effected on The Nasdaq Capital Market or otherwise and, if commenced, may be discontinued at any time.
In connection with this offering, the underwriter and selling group members may also engage in passive market making transactions in our common shares on The Nasdaq Capital Market. Passive market making consists of displaying bids on The Nasdaq Capital Market limited by the prices of independent market makers and effecting purchases limited by those prices in response to order flow. Rule 103 of Regulation M promulgated by the SEC limits the amount of net purchases that each passive market maker may make and the displayed size of each bid. Passive market making may stabilize the market price of our common shares at a level above that which might otherwise prevail in the open market and, if commenced, may be discontinued at any time.
Neither we nor the underwriter make any representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the price of our common shares. In addition, neither we nor the underwriter make any representation that the underwriter will engage in these transactions or that any transaction, if commenced, will not be discontinued without notice.
Electronic Offer, Sale and Distribution Of Shares
The underwriter or syndicate members may facilitate the marketing of this offering online directly or through one of their respective affiliates. In those cases, prospective investors may view offering terms and a prospectus online and place orders online or through their financial advisors. Such websites and the information contained on such websites, or connected to such sites, are not incorporated into and are not a part of this prospectus.
Other Relationships
The underwriter and its affiliates are full service financial institutions engaged in various activities, which may include securities trading, commercial and investment banking, financial advisory, investment management, investment research, principal investment, hedging, financing and brokerage activities. The underwriter has in the past, and may in the future, engage in investment banking and other commercial dealings in the ordinary course of business with us or our affiliates. The underwriter has in the past, and may in the future, receive customary fees and commissions for these transactions.
In the ordinary course of their various business activities, the underwriter and its affiliates may make or hold a broad array of investments and actively trade debt and equity securities (or related derivative securities) and financial instruments (including bank loans) for their own account and for the accounts of their customers, and such investment and securities activities may involve securities and/or instruments of the issuer. The underwriter and its affiliates may also make investment recommendations and/or publish or express independent research views in respect of such securities or instruments and may at any time hold, or recommend to clients that it acquires, long and/or short positions in such securities and instruments.
Listing
Our common shares trade in Canada on the TSX Venture Exchange under the trading symbol “DMA” and over-the-counter in the United States on the OTCQB marketplace under the trading symbol “DMCAF.” We have applied to list our common shares on The Nasdaq Capital Market under the trading symbol “DMAC.”
Transfer Agent and Registrar
The transfer agent and registrar for our common shares is Computershare Trust Company.
Selling Restrictions
Canada
The securities will not be qualified for distribution pursuant to a prospectus filed with the securities regulatory authorities in any of the provinces or territories of Canada and may not be offered or sold in Canada except on a private placement basis pursuant to an exemption from the prospectus requirements of applicable Canadian securities laws.
European Economic Area
In relation to each Member State of the European Economic Area which has implemented the Prospectus Directive, each, a Relevant Member State, an offer to the public of any of our common shares may not be made in that Relevant Member State, except that an offer to the public in that Relevant Member State of any of our common shares may be made at any time under the following exemptions under the Prospectus Directive, if they have been implemented in that Relevant Member State:
|
● |
to any legal entity which is a qualified investor as defined in the Prospectus Directive; |
|
● |
to fewer than 100 or, if the Relevant Member State has implemented the relevant provision of the 2010 PD Amending Directive, 150, natural or legal persons (other than qualified investors as defined in the Prospectus Directive), as permitted under the Prospectus Directive, subject to obtaining the prior consent of the representatives for any such offer; or |
|
● |
in any other circumstances falling within Article 3(2) of the Prospectus Directive, provided that no such offer of our common shares shall result in a requirement for the publication by us or any underwriter of a prospectus pursuant to Article 3 of the Prospectus Directive. |
For the purposes of this provision, the expression an “offer to the public” in relation to any of our common shares in any Relevant Member State means the communication in any form and by any means of sufficient information on the terms of the offer and any of our common shares to be offered so as to enable an investor to decide to purchase any of our common shares, as the same may be varied in that Member State by any measure implementing the Prospectus Directive in that Member State, the expression “Prospectus Directive” means Directive 2003/71/EC (and amendments thereto, including the 2010 PD Amending Directive, to the extent implemented in the Relevant Member State), and includes any relevant implementing measure in the Relevant Member State, and the expression “2010 PD Amending Directive” means Directive 2010/73/EU.
United Kingdom
The underwriter has represented and agreed that:
|
● |
it has only communicated or caused to be communicated and will only communicate or cause to be communicated an invitation or inducement to engage in investment activity (within the meaning of Section 21 of the Financial Services and Markets Act 2000 (“FSMA”)) received by it in connection with the issue or sale of our common shares in circumstances in which Section 21(1) of the FSMA does not apply to us; and |
|
● |
it has complied and will comply with all applicable provisions of the FSMA with respect to anything done by it in relation to our common shares in, from or otherwise involving the United Kingdom. |
Switzerland
The shares may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange (“SIX”), or on any other stock exchange or regulated trading facility in Switzerland. This document has been prepared without regard to the disclosure standards for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. Neither this document nor any other offering or marketing material relating to the shares or the offering may be publicly distributed or otherwise made publicly available in Switzerland.
Neither this document nor any other offering or marketing material relating to the offering, or the shares have been or will be filed with or approved by any Swiss regulatory authority. In particular, this document will not be filed with, and the offer of shares will not be supervised by, the Swiss Financial Market Supervisory Authority FINMA, and the offer of shares has not been and will not be authorized under the Swiss Federal Act on Collective Investment Schemes (“CISA”). Accordingly, no public distribution, offering or advertising, as defined in CISA, its implementing ordinances and notices, and no distribution to any non-qualified investor, as defined in CISA, its implementing ordinances and notices, shall be undertaken in or from Switzerland, and the investor protection afforded to acquirers of interests in collective investment schemes under CISA does not extend to acquirers of shares.
Australia
No placement document, prospectus, product disclosure statement or other disclosure document has been lodged with the Australian Securities and Investments Commission (“ASIC”), in relation to the offering.
This prospectus does not constitute a prospectus, product disclosure statement or other disclosure document under the Corporations Act 2001 (the “Corporations Act”), and does not purport to include the information required for a prospectus, product disclosure statement or other disclosure document under the Corporations Act.
Any offer in Australia of the shares may only be made to persons, the Exempt Investors, who are “sophisticated investors” (within the meaning of section 708(8) of the Corporations Act), “professional investors” (within the meaning of section 708(11) of the Corporations Act) or otherwise pursuant to one or more exemptions contained in section 708 of the Corporations Act so that it is lawful to offer the shares without disclosure to investors under Chapter 6D of the Corporations Act.
The shares applied for by Exempt Investors in Australia must not be offered for sale in Australia in the period of 12 months after the date of allotment under the offering, except in circumstances where disclosure to investors under Chapter 6D of the Corporations Act would not be required pursuant to an exemption under section 708 of the Corporations Act or otherwise or where the offer is pursuant to a disclosure document which complies with Chapter 6D of the Corporations Act. Any person acquiring shares must observe such Australian on-sale restrictions.
This prospectus contains general information only and does not take account of the investment objectives, financial situation or particular needs of any particular person. It does not contain any securities recommendations or financial product advice. Before making an investment decision, investors need to consider whether the information in this prospectus is appropriate to their needs, objectives and circumstances, and, if necessary, seek expert advice on those matters.
Israel
In the State of Israel this prospectus shall not be regarded as an offer to the public to purchase securities under the Israeli Securities Law, 5728 - 1968, which requires a prospectus to be published and authorized by the Israel Securities Authority, if it complies with certain provisions of Section 15 of the Israeli Securities Law, 5728 - 1968, including, inter alia, if: (i) the offer is made, distributed or directed to not more than 35 investors, subject to certain conditions, or the Addressed Investors; or (ii) the offer is made, distributed or directed to certain qualified investors defined in the First Addendum of the Israeli Securities Law, 5728 - 1968, subject to certain conditions, or the Qualified Investors. The Qualified Investors shall not be taken into account in the count of the Addressed Investors and may be offered to purchase securities in addition to the 35 Addressed Investors. Our company has not and will not take any action that would require it to publish a prospectus in accordance with and subject to the Israeli Securities Law, 5728 - 1968. We have not and will not distribute this prospectus or make, distribute or direct an offer to subscribe for our securities to any person within the State of Israel, other than to Qualified Investors and up to 35 Addressed Investors.
Qualified Investors may have to submit written evidence that they meet the definitions set out in of the First Addendum to the Israeli Securities Law, 5728 - 1968. In particular, we may request, as a condition to be offered securities, that Qualified Investors will each represent, warrant and certify to us or to anyone acting on our behalf: (i) that it is an investor falling within one of the categories listed in the First Addendum to the Israeli Securities Law, 5728 - 1968; (ii) which of the categories listed in the First Addendum to the Israeli Securities Law, 5728 - 1968 regarding Qualified Investors is applicable to it; (iii) that it will abide by all provisions set forth in the Israeli Securities Law, 5728 - 1968 and the regulations promulgated thereunder in connection with the offer to be issued securities; (iv) that the securities that it will be issued are, subject to exemptions available under the Israeli Securities Law, 5728 - 1968: (a) for its own account; (b) for investment purposes only; and (c) not issued with a view to resale within the State of Israel, other than in accordance with the provisions of the Israeli Securities Law, 5728 - 1968; and (v) that it is willing to provide further evidence of its Qualified Investor status. Addressed Investors may have to submit written evidence in respect of their identity and may have to sign and submit a declaration containing, inter alia, the Addressed Investor’s name, address and passport number or Israeli identification number
LEGAL MATTERS
The validity of the common shares being offered by this prospectus will be passed upon for us by Pushor Mitchell LLP, Kelowna, British Columbia, Canada. Certain legal matters relating to this offering will be passed upon for us by Fox Rothschild LLP, Minneapolis, Minnesota. Certain legal matters relating to this offering will be passed upon for the underwriter by Faegre Baker Daniels LLP, Minneapolis, Minnesota.
EXPERTS
The consolidated financial statements for the years ended December 31, 2017 and 2016 included in this prospectus have been audited by Baker Tilly Virchow Krause, LLP, our independent registered public accounting firm, and have been included herein in reliance upon the report of such firm given upon authority as experts in accounting and auditing.
WHERE YOU CAN FIND ADDITIONAL INFORMATION
We have filed with the SEC a registration statement on Form S-1 under the Securities Act with respect to our common shares offered by this prospectus. This prospectus, which constitutes part of that registration statement, does not contain all of the information set forth in the registration statement or the accompanying exhibits and schedules. Some items included in the registration statement are omitted from this prospectus in accordance with the rules and regulations of the SEC. For further information with respect to us and the common shares offered in this prospectus, we refer you to the registration statement and the accompanying exhibits and schedules. Statements contained in this prospectus regarding the contents of any contract, agreement or any other document are summaries of the material terms of these contracts, agreements or other documents. With respect to each of these contracts, agreements or other documents filed as an exhibit to the registration statement, reference is made to such exhibit for a more complete description of the matter involved.
A copy of the registration statement and the accompanying exhibits and schedules and any other document we file may be inspected without charge and copied at the SEC’s public reference room at 100 F Street, N.E., Washington, D.C. 20549. The public may obtain information on the operation of the public reference room by calling the SEC at 1-800-SEC-0330. The SEC maintains a website that contains reports, proxy and information statements and other information regarding registrants that file electronically with the SEC. The address of the SEC’s website is http://www.sec.gov.
In connection with this offering, we have registered our common shares with the SEC under Section 12(b) of the Exchange Act and have become subject to the information and periodic reporting requirements of the Exchange Act, and file periodic reports, proxy statements and other information with the SEC. These periodic reports, proxy statements and other information will be available for inspection and copying at the public reference room and website of the SEC referred to above. We maintain a website at http://www.diamedica.com. You may access our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports, proxy statements and other information filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act with the SEC free of charge at our website as soon as reasonably practicable after such material is electronically filed with, or furnished to, the SEC. The information contained in, or that can be accessed through, our website is not part of this prospectus.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
|
Audited Consolidated Financial Statements |
|
|
|
Page |
|
Report of Independent Registered Public Accounting Firm |
F-2 |
|
Consolidated Balance Sheets as of December 31, 2017 and 2016 |
F-3 |
|
Consolidated Statements of Operations and Comprehensive Loss for the years ended December 31, 2017 and 2016 |
F-4 |
|
Consolidated Statements of Stockholders’ Equity (Deficit) for the years ended December 31, 2017 and 2016 |
F-5 |
|
Consolidated Statements of Cash Flows for the years ended December 31, 2017 and 2016 |
F-6 |
|
Notes to Consolidated Financial Statements |
F-7 |
|
Unaudited Condensed Consolidated Financial Statements for the Three and Nine Months Ended September 30, 2018 and 2017 |
|
|
|
Page |
|
Condensed Consolidated Balance Sheets as of September 30, 2018 and December 31, 2017 |
F-22 |
|
Condensed Consolidated Statements of Operations and Comprehensive Loss |
F-23 |
|
Condensed Consolidated Statements of Stockholders’ Equity |
F-24 |
|
Condensed Consolidated Statements of Cash Flows |
F-25 |
|
Notes to Condensed Consolidated Financial Statements |
F-26 |
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of
DiaMedica Therapeutics Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of DiaMedica Therapeutics Inc. and Subsidiaries (the “Company”) as of December 31, 2017 and 2016, and the related consolidated statements of operations and comprehensive loss, stockholders’ deficit, and cash flows for each of the years then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2017 and 2016, and the results of its operations and its cash flows for each of the years then ended in conformity with accounting principles generally accepted in the United States of America.
Substantial Doubt about the Company’s Ability to Continue as a Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has recurring losses and negative cash flows from operations that raise substantial doubt about its ability to continue as a going concern. Management’s evaluations of the events and conditions and management’s plans regarding those matters are also described in Note 3. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board of the United States of America (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Baker Tilly Virchow Krause, LLP
We have served as the Company’s auditors since 2016.
Minneapolis, MN
August 27, 2018
DiaMedica Therapeutics Inc.
Consolidated Balance Sheets
(In thousands, except share amounts)
|
December 31, |
||||||||
|
2017 |
2016 |
|||||||
|
ASSETS |
||||||||
|
Current assets: |
||||||||
|
Cash |
$ | 1,353 | $ | 1,736 | ||||
|
Amounts receivable |
80 | 53 | ||||||
|
Prepaid expenses |
61 | 67 | ||||||
|
Total current assets |
1,494 | 1,856 | ||||||
|
Deposit |
271 | — | ||||||
|
Property and equipment, net |
37 | 19 | ||||||
|
Total non-current assets |
308 | 19 | ||||||
|
Total assets |
$ | 1,802 | $ | 1,875 | ||||
|
LIABILITIES AND STOCKHOLDERS’ EQUITY |
||||||||
|
Current liabilities: |
||||||||
|
Accounts payable and accrued liabilities |
$ | 919 | $ | 671 | ||||
|
Warrant liability |
84 | 93 | ||||||
|
Total current liabilities |
1,003 | 764 | ||||||
|
Commitments and contingencies (Note 10) |
||||||||
|
Stockholders’ equity: |
||||||||
|
Common shares, no par value; unlimited authorized; 6,370,664 and 5,526,049 shares issued and outstanding, as of December 31, 2017 and 2016, respectively |
— | — | ||||||
|
Additional paid-in capital |
41,033 | 37,085 | ||||||
|
Accumulated deficit |
(40,234 | ) | (35,974 | ) | ||||
|
Total stockholders’ equity |
799 | 1,111 | ||||||
|
Total liabilities and stockholders’ equity |
$ | 1,802 | $ | 1,875 | ||||
See accompanying notes to consolidated financial statements.
DiaMedica Therapeutics Inc.
Consolidated Statements of Operations and Comprehensive Loss
(In thousands, except share and per share amounts)
|
Year Ended December 31, |
||||||||
|
2017 |
2016 |
|||||||
|
Operating expenses: |
||||||||
|
Research and development |
$ | 3,206 | $ | 1,728 | ||||
|
General and administrative |
1,313 | 598 | ||||||
|
Operating loss |
(4,519 | ) | (2,326 | ) | ||||
|
Other (income) expense: |
||||||||
|
Governmental assistance - research incentives |
(244 | ) | — | |||||
|
Other (income) expense |
(6 | ) | 82 | |||||
|
Change in fair value of warrant liability |
(9 | ) | (188 | ) | ||||
|
Total other income |
(259 | ) | (106 | ) | ||||
|
Loss before income tax expense |
(4,260 | ) | (2,220 | ) | ||||
|
Income tax expense |
— | — | ||||||
|
Net loss and comprehensive loss |
$ | (4,260 | ) | $ | (2,220 | ) | ||
|
Basic and diluted net loss per share |
$ | (0.72 | ) | $ | (0.47 | ) | ||
|
Weighted average shares outstanding – basic and diluted |
5,935,790 | 4,735,751 | ||||||
See accompanying notes to consolidated financial statements.
DiaMedica Therapeutics Inc.
Consolidated Statements of Stockholders’ Equity (Deficit)
(In thousands except share amounts)
|
Common |
Additional |
Accumulated |
Total |
|||||||||||||
|
Balances at December 31, 2015 |
4,113,772 | $ | 32,576 | $ | (33,754 | ) | $ | (1,178 | ) | |||||||
|
Issuance of common shares and warrants, net of offering costs of $395 |
1,000,000 | 3,605 | — | 3,605 | ||||||||||||
|
Issuance of common shares and warrants, net of offering costs of $311 |
234,375 | 237 | — | 237 | ||||||||||||
|
Issuance of common shares in settlement of debt |
2,500 | 8 | — | 8 | ||||||||||||
|
Exercise of common share warrants |
174,108 | 442 | — | 442 | ||||||||||||
|
Issuance of common shares, deferred stock unit redemption |
1,294 | — | — | — | ||||||||||||
|
Share-based compensation expense |
— | 217 | — | 217 | ||||||||||||
|
Net loss |
— | — | (2,220 | ) | (2,220 | ) | ||||||||||
|
Balances at December 31, 2016 |
5,526,049 | $ | 37,085 | $ | (35,974 | ) | $ | 1,111 | ||||||||
|
Issuance of common shares and warrants, net of offering costs of $292 |
707,536 | 2,917 | — | 2,917 | ||||||||||||
|
Exercise of common share purchase warrants |
134,079 | 615 | — | 615 | ||||||||||||
|
Exercise of common share options |
3,000 | 7 | — | 7 | ||||||||||||
|
Share-based compensation expense |
— | 409 | — | 409 | ||||||||||||
|
Net loss |
— | — | (4,260 | ) | (4,260 | ) | ||||||||||
|
Balances at December 31, 2017 |
6,370,664 | $ | 41,033 | $ | (40,234 | ) | $ | 799 | ||||||||
See accompanying notes to consolidated financial statements.
DiaMedica Therapeutics Inc.
Consolidated Statements of Cash Flows
(In thousands)
|
Year Ended December 31, |
||||||||
|
2017 |
2016 |
|||||||
|
Cash flows from operating activities: |
||||||||
|
Net loss |
$ | (4,260 | ) | $ | (2,220 | ) | ||
|
Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||
|
Share-based compensation |
409 | 217 | ||||||
|
Change in fair value of warrant liability |
(9 | ) | (188 | ) | ||||
|
Depreciation |
4 | 2 | ||||||
|
Changes in operating assets and liabilities: |
||||||||
|
Amounts receivable |
(27 | ) | (44 | ) | ||||
|
Prepaid expenses |
6 | (33 | ) | |||||
|
Deposits |
(271 | ) | — | |||||
|
Accounts payable and accrued liabilities |
248 | (510 | ) | |||||
|
Deferred revenue |
— | (39 | ) | |||||
|
Other liabilities |
— | (172 | ) | |||||
|
Net cash used in operating activities |
(3,900 | ) | (2,987 | ) | ||||
|
Cash flows from investing activities: |
||||||||
|
Purchase of property and equipment |
(22 | ) | (7 | ) | ||||
|
Net cash used in financing activities |
(22 | ) | (7 | ) | ||||
|
Cash flows from financing activities: |
||||||||
|
Proceeds from issuance of common shares and warrants, net of offering costs |
2,917 | 517 | ||||||
|
Proceeds from issuance of common shares, net of offering costs |
— | 3,605 | ||||||
|
Proceeds from the exercise of common share purchase warrants |
615 | 442 | ||||||
|
Proceeds from the exercise of stock options |
7 | — | ||||||
|
Net cash provided by financing activities |
3,539 | 4,564 | ||||||
|
Net (decrease) increase in cash |
(383 | ) | 1,570 | |||||
|
Cash at beginning of year |
1,736 | 166 | ||||||
|
Cash at end of year |
$ | 1,353 | $ | 1,736 | ||||
|
Supplemental disclosure of non-cash transactions: |
||||||||
|
Common share purchase warrants issued as agent consideration |
$ | — | $ | 24 | ||||
|
Common shares issued in settlement of debt |
$ | — | $ | 8 | ||||
See accompanying notes to consolidated financial statements.
DiaMedica Therapeutics Inc.
Notes to Consolidated Financial Statements
|
1. |
Business |
DiaMedica Therapeutics Inc. and its wholly-owned subsidiaries, DiaMedica USA, Inc. and DiaMedica Australia Pty Ltd. (collectively “we,” “us,” “our” and the “Company”), exist for the primary purpose of advancing the clinical and commercial development of a proprietary recombinant KLK1 protein for the treatment of acute ischemic stroke and chronic kidney disease.
The Company is a listed company governed by the Canada Business Corporations Act and domiciled in British Columbia, Canada, whose shares are publicly traded on the TSX Venture Exchange in Canada under the symbol “DMA” and the OTCQB in the United States under the symbol “DMCAF.” The Company’s registered office is at 301 – 1665 Ellis Street, Kelowna, British Columbia V1Y 2B3. DiaMedica USA Inc. was incorporated under the laws of the State of Delaware on May 15, 2012. DiaMedica Australia Pty Ltd. was established on July 11, 2016 and incorporated under the laws of Australian Securities and Investments Commission.
Effective November 15, 2018, we implemented a 1-for-20 consolidation of our common shares. As previously announced, the share consolidation was approved by our stockholders as of November 6, 2018 and was intended to increase the market price per share of our common stock to a level that qualifies for listing on the Nasdaq Capital Market. All references to share and per share amounts included in these consolidated financial statements have been retroactively restated to reflect the share consolidation. See Note 14 titled “Subsequent Events” for additional information.
|
2. |
Risks, Uncertainties and Going Concern |
The Company operates in a highly regulated and competitive environment. The development, manufacturing and marketing of pharmaceutical products require approval from, and are subject to ongoing oversight by, the Food and Drug Administration (“FDA”) in the United States, the Therapeutic Goods Administration (“TGA”) in Australia, the European Medicines Agency (“EMA”) in the European Union, and comparable agencies in other countries. Obtaining approval for a new pharmaceutical product is never certain, may take many years, and is normally expected to involve substantial expenditures.
As of December 31, 2017, we have incurred losses of $40.2 million since our inception in 2000. For the year ended December 31, 2017, we incurred a net loss and negative cash flows from operating activities of $4.3 million and $3.9 million, respectively. We expect to incur substantial losses for the foreseeable future, which will continue to generate negative net cash flows from operating activities, as we continue to pursue increasing levels of research and development activities and the clinical development of our primary product candidate, DM199. As of December 31, 2017, we had cash of $1.4 million, working capital of $491,000 and stockholders’ equity of $799,000. The Company’s principal sources of cash have included the issuance of equity securities.
The accompanying consolidated financial statements have been prepared assuming that we will continue as a going concern which contemplates the realization of assets and liquidation of liabilities in the normal course of business and do not include any adjustments relating to the recoverability or classification of assets or the amounts of liabilities that might result from the outcome of these uncertainties. Our ability to continue as a going concern, realize the carrying value of our assets and discharge our liabilities in the ordinary course of business is dependent upon a number of factors, including our ability to obtain additional financing, the success of our development efforts, our ability to obtain marketing approval for our initial product candidate, DM199, in the United States, Australia, the European Union or other markets and ultimately our ability to market and sell our initial product candidate. These factors, among others, raise substantial doubt about our ability to continue operations as a going concern. See Note 3 titled “Liquidity, Management’s Plans and Going Concern.”
|
3. |
Liquidity and Management Plans |
As of December 31, 2017 and March 31, 2018, the Company has an accumulated deficit of $40.2 million and $40.9 million, respectively, and the Company has not generated positive cash flow from operations since its inception.
Additional funding will be required to continue the Company’s research and development and other operating activities. In the next 12 months we will likely seek to raise additional funds through various sources, such as equity and debt financings, or through strategic collaborations and license agreements. We can give no assurances that we will be able to secure additional sources of funds to support our operations, or if such funds are available to us, that such additional financing will be sufficient to meet our needs or on terms acceptable to us. This risk would increase if our clinical data is not positive or economic and market conditions deteriorate.
During March 2018, the Company completed a brokered and non-brokered private placement of 1,322,965 units at a price of $4.90 per unit for aggregate gross proceeds of approximately $6.3 million. In addition, during February 2018, 121,256 common shares were issued on the exercise of warrants for gross proceeds of approximately $484,000. See Note 14 titled “Subsequent Events” for further details.
If we are unable to obtain additional financing when needed, we would need to scale back our operations taking actions that may include, among other things, reducing use of outside professional service providers, reducing staff or staff compensation, significantly modify or delay the development of our DM199 product candidate, license to third parties the rights to commercialize our DM199 product candidate for acute ischemic stroke, chronic kidney disease or other applications that we would otherwise seek to pursue, or cease operations.
Our future success is dependent upon our ability to obtain additional financing, the success of our development efforts, our ability to demonstrate clinical progress for our DM199 product candidate in the United States or other markets, our ability to obtain required governmental approvals of our product candidate and ultimately our ability to license or market and sell our DM199 product candidate. If we are unable to obtain additional financing when needed, if our clinical trials are not successful, if we are unable to obtain required governmental approvals, we would not be able to continue as a going concern and would be forced to cease operations and liquidate our company.
There can be no assurances that we will be able to obtain additional financing on commercially reasonable terms, or at all. The sale of additional equity securities would likely result in dilution to our current stockholders.
|
4. |
Summary of Significant Accounting Policies |
Basis of presentation
We have prepared the accompanying consolidated financial statements in conformity with accounting principles generally accepted in the United States of America (“GAAP”) which contemplates the realization of its assets and the settlement of its liabilities in the normal course of operations. Our fiscal year ends on December 31.
Principles of consolidation
The accompanying consolidated financial statements include the assets, liabilities and expenses of DiaMedica Therapeutics Inc. and our wholly-owned subsidiaries. All significant intercompany transactions and balances have been eliminated in consolidation.
Functional currency
The United States dollar is the functional currency that represents the economic effects of the underlying transactions, events and conditions and various other factors including the currency of historical and future expenditures and the currency in which funds from financing activities are mostly generated by the Company. A change in the functional currency occurs only when there is a material change in the underlying transactions, events and condition. A change in functional currency could result in material differences in the amounts recorded in the consolidated statement of loss and comprehensive loss for foreign exchange gains and losses. All amounts in the accompanying consolidated financial statements are in U.S. dollars unless otherwise indicated.
Use of estimates
The preparation of consolidated financial statements requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amount of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Concentration of credit risk
Financial instruments that potentially subject the company to significant concentrations of credit risk consist primarily of cash. Cash is deposited in demand accounts at commercial banks. At times, such deposits may be in excess of insured limits. The Company has not experienced any losses on its deposits of cash.
Fair value of financial instruments
Carrying amounts of certain of the Company’s financial instruments, including amounts receivable, prepaid expenses and other current assets, accounts payable and accrued liabilities approximate fair value due to their short maturities. Certain of the Company’s common share purchase warrants are required to be reported at fair value. The fair value of common share purchase warrants is disclosed in Note 9 titled “Warrant Liability.”
Fair value measurements
Fair value is defined as the exit price, or the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants as of the measurement date. The authoritative guidance also establishes a hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. Observable inputs are inputs market participants would use in valuing the asset or liability developed based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company’s assumptions about the factors market participants would use in valuing the asset or liability developed based upon the best information available in the circumstances. The categorization of financial assets and financial liabilities within the valuation hierarchy is based upon the lowest level of input that is significant to the fair value measurement.
The hierarchy is broken down into three levels defined as follows:
|
● |
Level 1—Unadjusted quoted prices in active markets that are accessible at the measurement date for identical, unrestricted assets or liabilities. |
|
● |
Level 2—Quoted prices for similar assets and liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets that are not active or model-derived valuations for which all significant inputs are observable, either directly or indirectly. |
|
● |
Level 3—Prices or valuation techniques that require inputs that are both significant to the fair value measurement and unobservable. |
Our cash consists of bank deposits. As of December 31, 2017, the Company believes that the carrying amounts of its other financial instruments, including amounts receivable, accounts payable and accrued liabilities, approximate their fair value due to the short-term maturities of these instruments.
Common share warrant liability
The common share warrants that were issued in connection with the February 2016 private placements of common shares are classified as a liability in the consolidated balance sheets, as the common share warrants have an exercise price stated in Canadian dollars, which is different than the functional currency, and thus these warrants qualify as a derivative instruments. The fair value of these common share warrants is re-measured at each financial reporting period and immediately before exercise, with any changes in fair value being recognized as a component of other income (expense) in the consolidated statements of operations.
Long-lived assets
Property and equipment are stated at purchased cost less accumulated depreciation. Depreciation of property and equipment is computed using the straight-line method over their estimated useful lives of three to ten years for office equipment and four years for computer equipment. Upon retirement or sale, the cost and related accumulated depreciation are removed from the consolidated balance sheets and the resulting gain or loss is reflected in the consolidated statements of operations. Repairs and maintenance are expensed as incurred.
Long-lived assets are evaluated for impairment when events or changes in circumstances indicate that the carrying amount of the asset or related group of assets may not be recoverable. If the expected future undiscounted cash flows are less than the carrying amount of the asset, an impairment loss is recognized at that time. Measurement of impairment may be based upon appraisal, market value of similar assets or discounted cash flows.
Research and development costs
Research and development costs include expenses incurred in the conduct of human clinical trials, for third-party service providers performing various testing and accumulating data related to non-clinical studies; sponsored research agreements; developing the manufacturing process necessary to produce sufficient amounts of the DM199 compound for use in our non-clinical studies and human clinical trials; consulting resources with specialized expertise related to execution of our development plan for our DM199 product candidate; and personnel costs, including salaries, benefits and share-based compensation.
We charge research and development costs, including clinical trial costs, to expense when incurred. Our human clinical trials are performed at clinical trial sites and are administered jointly by us with assistance from contract research organizations (“CROs”). Costs of setting up clinical trial sites are accrued upon execution of the study agreement. Expenses related to the performance of clinical trials are accrued based on contracted amounts and the achievement of agreed upon milestones, such as patient enrollment, patient follow-up, etc. We monitor levels of performance under each significant contract, including the extent of patient enrollment and other activities through communications with the clinical trial sites and CROs, and adjust the estimates, if required, on a quarterly basis so that clinical expenses reflect the actual work performed at each clinical trial site and by each CRO.
Patent costs
Costs associated with prosecuting and maintaining patents are expensed as incurred given the uncertainty of patent approval and, if approved, resulting in probable future economic benefit to the Company. Patent-related costs, including legal expenses, included in research and development costs were $160,000 and $45,000 for the years ended December 31, 2017 and 2016, respectively.
Share-based compensation
The cost of employee and non-employee services received in exchange for awards of equity instruments is measured and recognized based on the estimated grant date fair value of those awards. Compensation cost is recognized ratably using the straight-line attribution method over the vesting period, which is considered to be the requisite service period. We record forfeitures in the periods in which they occur.
The fair value of share-based awards is estimated using the Black-Scholes option pricing model. The determination of the fair value of share-based awards is affected by our stock price, as well as assumptions regarding a number of complex and subjective variables. Risk free interest rates are based upon Canadian Government bond rates appropriate for the expected term of each award. Expected volatility rates are based on the on historical volatility equal to the expected life of the option. The assumed dividend yield is zero, as we do not expect to declare any dividends in the foreseeable future. The expected term of options is estimated considering the vesting period at the grant date, the life of the option and the average length of time similar grants have remained outstanding in the past.
Income taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the Consolidated Financial Statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carry-forwards. Deferred tax assets and liabilities are measured using enacted rates, for each of the jurisdictions in which the Company operates, expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in operations in the period that includes the enactment date. Valuation allowances are established when necessary to reduce deferred tax assets to the amount that is more likely than not to be realized. The Company has provided a full valuation allowance against the gross deferred tax assets as of December 31, 2017 and 2016. See Note 13, “Income Taxes” for additional information. The Company’s policy is to classify interest and penalties related to income taxes as income tax expense in the Consolidated Statements of Operations and Comprehensive Loss.
Government assistance
Government assistance relating to research and development performed by DiaMedica Australia Pty Ltd. is recorded as a component of Other (Income) Expense. Government assistance is initially recognized when reasonable assurance exists that the Company will comply with the conditions attached to the incentive program and that the incentive payments will be received. In subsequent periods, the government assistance is recognized when the related expenditures are incurred.
Net loss per share
We compute net loss per share by dividing our net loss (the numerator) by the weighted-average number of common shares outstanding (the denominator) during the period. Shares issued during the period and shares reacquired during the period, if any, are weighted for the portion of the period that they were outstanding. The computation of diluted earnings per share, or diluted EPS, is similar to the computation of basic EPS except that the denominator is increased to include the number of additional common shares that would have been outstanding if the dilutive potential common shares had been issued. Our diluted EPS is the same as basic EPS due to common equivalent shares being excluded from the calculation, as their effect is anti-dilutive.
The following table summarizes our calculation of net loss per common share for the periods (in thousands, except share and per share data):
|
December 31, |
||||||||
|
2017 |
2016 |
|||||||
|
Net loss |
$ | (4,260 | ) | $ | (2,220 | ) | ||
|
Weighted average shares outstanding—basic and diluted |
5,935,790 | 4,735,751 | ||||||
|
Basic and diluted net loss per share |
$ | (0.72 | ) | $ | (0.47 | ) | ||
The following outstanding potential common shares were not included in the diluted net loss per share calculations as their effects were not dilutive:
|
Year Ended December 31, |
||||||||
|
2017 |
2016 |
|||||||
|
Employee and non-employee stock options |
480,035 | 425,350 | ||||||
|
Common shares issuable under common share purchase warrants |
216,213 | 128,103 | ||||||
|
Common shares issuable under deferred share unit plan |
21,183 | 21,183 | ||||||
| 717,431 | 574,636 | |||||||
Recently issued accounting pronouncement
In February 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2016-02, Leases. The guidance in ASU 2016-02 supersedes the lease recognition requirements in the Accounting Standards Codification Topic 840, Leases. ASU 2016-02 requires an entity to recognize assets and liabilities arising from a lease for both financing and operating leases, along with additional qualitative and quantitative disclosures. The new standard requires the immediate recognition of all excess tax benefits and deficiencies in the income statement and requires classification of excess tax benefits as an operating activity as opposed to a financing activity in the statements of cash flows. ASU 2016-02 is effective for fiscal years beginning after December 15, 2018, with early adoption permitted. Management is evaluating the standard’s impact on the consolidated financial statements.
In June 2018, the FASB issued ASU 2018-07, Compensation – Stock Compensation (Topic 718): Improvements to Nonemployee Share-Based Payment Accounting, which simplifies several aspects of the accounting for nonemployee share-based payment transactions resulting from expanding the scope of Topic 718 to include share-based payment transactions for acquiring goods and services from nonemployees. This ASU is effective for the Company for fiscal years beginning after December 15, 2018, including interim periods within that fiscal year. Early adoption is permitted, but no earlier than an entity’s adoption date of Topic 606. The Company is currently evaluating the impact of the new guidance on our consolidated financial statements.
Recently adopted accounting pronouncements
In March 2016, the FASB issued ASU No. 2016-09, Improvements to Employee Share-Based Payment Accounting. The guidance in ASU 2016-09 is intended to simplify aspects of the accounting for employee share-based payments, including the accounting for income taxes, forfeitures, statutory withholding requirements, and classification on the statement of cash flows. The standard is effective for interim and annual periods beginning after December 15, 2016, with early adoption permitted. The adoption of ASU 2016-09 during the year ended December 31, 2016 did not have a material impact on the consolidated financial statements and related disclosures.
In July 2017, the FASB issued ASU 2017-11, Earnings Per Share (Topic 260), Distinguishing Liabilities from Equity (Topic 480) and Derivatives and Hedging (Topic 815): I. Accounting for Certain Financial Instruments with Down Round Features; and II. Replacement of the Indefinite Deferral for Mandatorily Redeemable Financial Instruments of Certain Nonpublic Entities and Certain Mandatorily Redeemable Non-controlling Interests with a Scope Exception. Part I of this update addresses the complexity of accounting for certain financial instruments with down round features. Down round features are features of certain equity-linked instruments (or embedded features) that result in the strike price being reduced on the basis of the pricing of future equity offerings. Current accounting guidance creates cost and complexity for entities that issue financial instruments (such as warrants and convertible instruments) with down round features that require fair value measurement of the entire instrument or conversion option. Part II of this update addresses the difficulty of navigating Topic 480, Distinguishing Liabilities from Equity, because of the existence of extensive pending content in the FASB Accounting Standards Codification. This pending content is the result of the indefinite deferral of accounting requirements about mandatorily redeemable financial instruments of certain nonpublic entities and certain mandatorily redeemable non-controlling interests. The amendments in Part II of this update do not have an accounting effect. This ASU is effective for fiscal years, and interim periods within those years, beginning after December 15, 2018, with early adoption permitted. The Company adopted ASU 2017-11 during the year ended December 31, 2017. Due to the adoption, the December 2017 warrants were not accounted for as derivative instruments. There was no activity in prior years which fall under this guidance. As such, early adoption has no effect on prior years.
|
5. |
Amounts Receivable |
Amounts receivable consisted of the following (in thousands):
|
December 31, 2017 |
December 31, 2016 |
|||||||
|
Sales-based taxes receivable |
80 | 53 | ||||||
|
Total amounts receivable |
$ | 80 | $ | 53 | ||||
|
6. |
Deposit |
Deposit consisted of the following (in thousands):
|
December 31, 2017 |
December 31, 2016 |
|||||||
|
Advances to vendor |
$ | 271 | $ | — | ||||
|
Total Deposit |
$ | 271 | $ | — | ||||
We have advanced funds to a vendor engaged to support the performance of the REMEDY Phase II clinical trial. The funds advanced will be held, interest free, by this vendor until the completion of the trial and applied to final trial invoices or refunded. This deposit is classified as non-current as the trial is not expected to be completed during 2018.
|
7. |
Property and Equipment |
Property and equipment consisted of the following (in thousands):
|
December 31, 2017 |
December 31, 2016 |
|||||||
|
Furniture and equipment |
$ | 40 | $ | 22 | ||||
|
Computer equipment |
23 | 20 | ||||||
| 63 | 42 | |||||||
|
Less accumulated depreciation |
(26 | ) | (23 | ) | ||||
|
Property and equipment, net |
$ | 37 | $ | 19 | ||||
|
8. |
Accrued Liabilities |
Accounts payable and accrued liabilities consisted of the following (in thousands):
|
December 31, 2017 |
December 31, 2016 |
|||||||
|
Trade and other payables |
$ | 513 | $ | 250 | ||||
|
Accrued compensation and related |
355 | 142 | ||||||
|
Accrued research and other professional fees |
45 | 255 | ||||||
|
Other accrued liabilities |
6 | 24 | ||||||
|
Total accrued liabilities |
$ | 919 | $ | 671 | ||||
|
9. |
Warrant Liability |
In February 2016, the Company completed, in two tranches, a non-brokered private placement of 234,375 units with each unit consisting of one common share and one half of one common share purchase warrant. The Company issued 117,188 warrants. Each warrant entitles the holder to purchase one common share at a price of $5.00 Canadian dollars at any time prior to expiry on February 18 or 25, 2018 for Tranche 1 and Tranche 2, respectively.
As the warrant exercise price is stated in Canadian dollars and the Company’s functional currency is the U.S. dollar, the warrants are deemed to be a derivative, with their estimated fair value classified as a liability on the Company’s balance sheet. The initial estimated fair value of the warrants was recorded as a warrant liability with subsequent changes in the estimated fair value recognized in the Consolidated Statements of Operations and Comprehensive Loss. The Company allocated $257,000 of the net proceeds to the warrant liability and the balance of the proceeds to the common shares (Note 9). The initial fair value of the warrants was determined using a Black-Scholes pricing model with the following assumptions: expected volatilities of 191.8 – 225.0%, risk-free interest rates of 0.43 – 0.49%, and expected life of 2 years.
In connection with the offering, the Company issued an aggregate of 10,915 compensation warrants. Each compensation warrant entitles the holder to purchase one common share at $5.00 Canadian dollars for a period of 2 years from the date of issuance, subject to acceleration on the same terms as the common share purchase warrants. The Company estimated the value of these warrants at $24,000, which was included in the issuance costs. The initial fair value of the warrants was determined using a Black-Scholes pricing model with the following assumptions: expected volatilities of 191.8 – 225.0%, risk-free interest rates of 0.43 – 0.49%, and expected life of 2 years.
The fair value of the Company’s common share purchase warrant liability, for both investor warrants and compensation warrants, is calculated using a Black-Scholes valuation model and is classified as Level 3 in the fair value hierarchy. The fair values were estimated using the following valuation assumptions:
|
Unit Warrants |
Compensation Warrants |
||||||
|
2017 |
2016 |
2017 |
2016 |
||||
|
Common share fair value |
$0.26 – $0.42 |
$0.16 – $0.24 |
$0.26 – $0.42 |
$0.16 – $0.24 |
|||
|
Risk-free interest rate |
0.75% – 1.67% |
0.43% – 0.76% |
0.75% – 1.67% |
0.43% – 0.76% |
|||
|
Expected dividend yield |
0% |
0% |
0% |
0% |
|||
|
Expected life (years) |
0.13 – 0.89 |
1.1 – 2.0 |
0.13 – 0.89 |
1.1 – 2.0 |
|||
|
Expected stock price volatility |
20.8% – 105.3% |
89.6% – 191.8% |
20.8% – 105.3% |
89.6% – 191.8% |
|||
The following is a rollforward of the fair value of Level 3 warrants (in thousands):
|
Warrant Liability |
||||
|
Warrant issuance – February 2016 |
$ | 281 | ||
|
Change in fair value |
(188 | ) | ||
|
Ending balance December 31, 2016 |
93 | |||
|
Change in fair value |
(9 | ) | ||
|
Ending balance December 31, 2017 |
$ | 84 | ||
|
10. |
Commitments and Contingencies |
Clinical trials and product development
In the normal course of business, the Company incurs obligations to make future payments as it executes its business plan. These contracts relate to preclinical, clinical and development activities, including the clinical research organization conducting our Phase II clinical trial for acute ischemic stroke. These commitments are subject to significant change and the ultimate amounts due may be materially different as these obligations are affected by, among other factors, the number and pace of patients enrolled, the number of clinical study sites, amount of time to complete study enrollments and the time required to finalize the analysis and reporting of study results. Clinical research agreements are generally cancelable upon 30 days’ notice, with the Company’s obligation then limited to costs incurred up to that date. Cancelation terms for product development contracts vary and are generally dependent upon timelines for sourcing research materials and reserving laboratory time. As of December 31, 2017, the Company estimates that its outstanding commitments including research and development contracts are approximately $2.2 million over the next 12 months and approximately $700,000 in the following 12 months.
On September 11, 2017, the Company announced the initiation of REMEDY, a 60-patient Phase II clinical trial evaluating DM199 in patients with acute ischemic stroke (“AIS”). The study drug (DM199 or placebo) will be administered as an intravenous (“IV”) infusion within 24 hours of stroke symptom onset, followed by subcutaneous (under the skin) injections once every 3 days for 21 days. The study is designed to measure safety and tolerability along with multiple tests designed to investigate DM199’s therapeutic potential including plasma-based biomarkers and standard functional stroke measures assessed at 90 days post-stroke (Modified Rankin Scale (“MRS”), National Institutes of Health Stroke Scale (NIHSS), Barthel Index (BI), and CRP, a measure of inflammation).
Additional clinical trials will be subsequently required if the results of the Phase II are positive. However, at this time, we are unable to reasonably estimate the total costs of future trials. Such costs are dependent upon and subject to change depending on the results of current and future clinical trials as well as developments in the regulatory requirements. Clinical trial costs are expensed as incurred.
Technology license
The Company has entered into a research, development, and license agreement whereby the Company receives research services and rights to proprietary technologies. Milestone and royalty payments that may become due under this agreement with such payments dependent upon, among other factors, clinical trials, regulatory approvals and ultimately the successful development of a new drug, the outcome and timing of which is uncertain. There were no amounts due or payable under this agreement during 2017 and 2016.
Indemnification of directors and officers
The Company, as permitted under laws of the Canada and in accordance with its by-laws, will indemnify and advance expenses to its directors and officers to the fullest extent permitted by law or, if applicable, pursuant to indemnification agreements. They further provide that we may choose to indemnify other employees or agents of our Company from time to time. The Company has secured insurance on behalf of any officer, director, employee or other agent for any liability arising out of his or her actions in connection with their services to the Company. As of December 31, 2017, there was no pending litigation or proceeding involving any director or officer of the Company as to which indemnification is required or permitted, and we are not aware of any threatened litigation or proceeding that may result in a claim for indemnification. Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the Company, the Company has been advised that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
The Company believes the fair value of these indemnification agreements is minimal. Accordingly, the Company had not recorded any liabilities for these obligations as of December 31, 2017 or 2016.
Future minimum lease payments
The Company leases certain office space under a non-cancelable operating lease. On May 3, 2017, the Company amended the lease agreement to extend its lease term by 42 months, for an expiration date of August 31, 2022, and increase its leased space. Rent is expensed on a straight-line basis.
Future minimum lease payment under this operating lease are as follows (in thousands):
|
2018 |
$ | 62 | ||
|
2019 |
64 | |||
|
2020 |
66 | |||
|
2021 |
68 | |||
|
2022 |
45 | |||
| $ | 305 |
|
11. |
Stockholders’ Deficit |
Authorized capital stock
The Company has authorized share capital of an unlimited number of common voting shares and the shares have no stated par value.
Common shareholders are entitled to receive dividends as declared by the Company, if any, and are entitled to one vote per share at the Company’s annual general meeting and any extraordinary general meeting.
Shareholders rights plan
The Company adopted a shareholder rights plan agreement (the “Rights Plan”). The Rights Plan is designed to provide adequate time for the Board of Directors and the shareholders to assess an unsolicited takeover bid for DiaMedica, to provide the Board of Directors with sufficient time to explore and develop alternatives for maximizing shareholder value if a takeover bid is made, and to provide shareholders with an equal opportunity to participate in a takeover bid and receive full and fair value for their common shares. The Rights Plan was renewed at the Company’s annual meeting of shareholders in December 2017 and is set to expire at the close of the Company’s annual meeting of shareholders in 2020.
The rights issued under the Rights Plan will initially attach to and trade with the common shares and no separate certificates will be issued unless an event triggering these rights occurs. The rights will become exercisable only when a person, including any party related to it, acquires or attempts to acquire 20 percent (20%) or more of the outstanding common shares without complying with the “Permitted Bid” provisions of the Rights Plan or without approval of the Board of Directors. Should such an acquisition occur or be announced, each right would, upon exercise, entitle a rights holder, other than the acquiring person and related persons, to purchase common shares at a 50 percent (50%) discount to the market price at the time.
Under the Plan, a Permitted Bid is a bid made to all holders of the common shares and which is open for acceptance for not less than sixty (60) days. If at the end of sixty (60) days at least 50 percent (50%) of the outstanding common shares, other than those owned by the offeror and certain related parties have been tendered, the offeror may take up and pay for the common shares but must extend the bid for a further ten (10) days to allow other shareholders to tender.
The issuance of common shares upon the exercise of the rights is subject to receipt of certain regulatory approvals.
Private placements during 2017
On December 18, 2017, the Company completed a non-brokered private placement of 181,220 units at a price of $5.20 per unit for aggregate gross proceeds of approximately $944,000, or $934,000 net of issuance costs. Each unit consisted of one common share and one half of one common share purchase warrant. Each whole warrant entitles the holder to purchase one common share at a price of $7.00 at any time prior to expiry on December 19, 2019. Warrants are subject to early expiry, at the option of the Company, if on any date the volume-weighted average closing trading price of the common shares on any recognized Canadian stock exchange equals or exceeds $12.00 for a period of 21 consecutive trading days.
On April 17, 2017, the Company completed a non-brokered private placement of 526,316 units at a price of $3.80 per unit for aggregate gross proceeds of approximately $2,000,000, or $1,983,000 net of issuance costs. Each unit consisted of one common share and one half of one common share purchase warrant. Each whole warrant entitles the holder to purchase one common share at a price of $4.60 at any time prior to expiry on April 17, 2019. Warrants are subject to early expiry, at the option of the Company, if on any date the volume-weighted average closing trading price of the common shares on any recognized Canadian stock exchange equals or exceeds $6.00 for a period of 10 consecutive trading days.
During the year ended December 31, 2017, 134,079 common shares were issued on the exercise of warrants for gross proceeds of $615,000 and 3,000 common shares were issued on the exercise of options for gross proceeds of $7,000.
Private placements during 2016
On August 22, 2016 and September 8, 2016, the Company completed a non-brokered private placement of 750,000 and 250,000 common shares, respectively, at a price of $4.00 per share for aggregate gross proceeds of $4,000,000, or $3,605,000 net of issuance costs.
On April 22, 2016, the Company issued 2,500 common shares for settlement of a debt to a vendor at an effective issue price of approximately $3.20 per common share.
On February 25, 2016, the Company completed the second tranche of a non-brokered private placement of 190,625 units at a price of $2.34 per unit for aggregate gross proceeds of approximately $102,000. Each unit consisted of one common share and one half of one common share purchase warrant. Each whole warrant entitles the holder to purchase one common share at a price of CAD$5.00 at any time prior to expiry of February 25, 2018. In connection with the financing, the Company issued 3,500 compensation warrants and paid a finder’s fee of 8% of the aggregate gross proceeds. Each compensation warrant entitles the holder to acquire one common share at an exercise price of CAD$5.00 prior to expiry on February 25, 2018.
The proceeds from the sale were allocated first to the warrants as a derivative liability and the remainder to the common shares. As a result, approximately $52,000 of the proceeds were allocated to the warrant derivative liability and the remaining proceeds of approximately $50,000, before offering costs, were allocated to the common shares.
On February 18, 2016, the Company completed the first tranche of a non-brokered private placement of 190,625 units at a price of $2.34 per unit for aggregate gross proceeds of approximately $446,000. Each unit consisted of one common share and one half of one common share purchase warrant. Each whole warrant entitles the holder to purchase one common share at a price of CAD$5.00 at any time prior to expiry on February 18, 2018. In connection with the financing, the Company issued 7,415 compensation warrants and paid a net finder’s fee of 4% of the aggregate gross proceeds. Each compensation warrant entitles the holder to acquire one common share at an exercise price of CAD$5.00 prior to expiry on February 18, 2018.
The proceeds from the sale were allocated first to the warrants as a derivative liability and the remainder to the common shares. As a result, approximately $205,000 of the proceeds were allocated to the warrant derivative liability and the remaining proceeds of approximately $240,000, before offering costs, were allocated to the common shares.
During the year ended December 31, 2016, 1,294 common shares were issued on the redemption of deferred share units and 174,108 common shares were issued on the exercise of warrants for gross proceeds of $442,000, and 544,554 warrants expired unexercised.
Shares reserved
Shares of common stock reserved for future issuance are as follows:
|
December 31, 2017 |
||||
|
Stock options outstanding |
480,035 | |||
|
Deferred share units outstanding |
21,183 | |||
|
Shares available for grant under the DiaMedica Stock Option Plan |
216,213 | |||
|
Common shares issuable under common stock purchase warrants |
157,032 | |||
|
Total |
874,463 | |||
|
12. |
Share-based Compensation |
Deferred share unit plan
The 2012 Deferred Share Unit Plan (the “2012 DSU Plan”) promotes greater alignment of long-term interests between non-executive directors and executive officers of the Company and its shareholders through the issuance of deferred share units (“DSUs”). Since the value of DSUs increases or decreases with the market price of the common shares, DSUs reflect a philosophy of aligning the interests of directors and executive officers by tying compensation to share price performance. For the years ended December 31, 2017 and 2016, there were zero and 18,750 shares issued, respectively, with an intrinsic value of zero and $53,000, respectively, for payment of directors’ fees. The Company has reserved for issuance up to 100,000 common shares under the 2012 DSU Plan and 21,183 DSUs were outstanding at December 31, 2017 and 2016.
Stock option plan
DiaMedica has adopted a Stock Option Plan (the “Option Plan”) where the Board of Directors may from time to time, in their sole discretion, and in accordance with the requirements of the Toronto (TSX) Venture Exchange, grant to directors, officers, management company employees, investor relations consultants and Consultants (as such terms are used in the Stock Option Plan) to DiaMedica, non-transferable options to purchase common shares. The shareholders approved the adoption of an Option Plan on September 22, 2011, and as amended and restated on October 23, 2015 and December 21, 2017, reserving for issuance up to 10% of the Company’s issued and outstanding common shares. Options granted vest at various rates and have terms of up to 10 years. As of December 31, 2017, options to purchase 480,035 common shares were outstanding. As the TSX Venture Exchange is the principal trading market for the Company’s shares, all options have been priced in Canadian dollars.
The aggregate number of common shares reserved as of December 31, 2017 was 637,050, which includes both the Option Plan and the 2012 DSU Plan.
We recognize share-based compensation based on the fair value of each award as estimated using the Black-Scholes option valuation model. Ultimately, the actual expense recognized over the vesting period will only be for those shares that actually vest.
A summary of option activity is as follows:
|
Shares |
Weighted Average |
Aggregate Intrinsic Value |
||||||||||
|
Balances at December 31, 2015 |
320,600 | $ | 9.85 | $ | 60,000 | |||||||
|
Shares Reserved |
— | — | ||||||||||
|
Granted |
138,750 | 4.83 | ||||||||||
|
Exercised |
— | — | ||||||||||
|
Expired / cancelled |
(24,000 | ) | 14.40 | |||||||||
|
Forfeited |
(7,500 | ) | 26.20 | |||||||||
|
Balances at December 31, 2016 |
427,850 | $ | 7.68 | $ | 187,120 | |||||||
|
Granted |
127,635 | 6.11 | ||||||||||
|
Exercised |
(3,000 | ) | 3.00 | |||||||||
|
Expired / cancelled |
(72,450 | ) | 13.29 | |||||||||
|
Forfeited |
— | — | ||||||||||
|
Balances at December 31, 2017 |
480,035 | $ | 6.45 | $ | 674,481 | |||||||
A summary of the status of our unvested shares during the year ended and as of December 31, 2017 is as follows:
|
Shares Under Option |
Weighted Average Grant Date Fair Value Per Share (CAD$) |
|||||||
|
Unvested at December 31, 2016 |
225,758 | $ | 2.66 | |||||
|
Granted |
127,635 | 4.78 | ||||||
|
Vested |
(125,400 | ) | 2.12 | |||||
|
Forfeitures |
(11,200 | ) | 3.08 | |||||
|
Unvested at December 31, 2017 |
216,793 | $ | 3.69 | |||||
Information about stock options outstanding, vested and expected to vest as of December 31, 2017, is as follows:
|
Outstanding, Vested and Expected to Vest |
Options Vested and Exercisable |
|||||||||||||||||||
|
Weighted Average |
Weighted |
Weighted Average |
||||||||||||||||||
|
Per Share |
Remaining |
Average |
Remaining |
|||||||||||||||||
|
Exercise Price |
Contractual |
Exercise Price |
Options |
Contractual |
||||||||||||||||
|
(CAD$) |
Shares |
Life (Years) |
(CAD$) |
Exercisable |
Life (Years) |
|||||||||||||||
|
$2.00 - $2.60 |
55,000 | 7.73 | $ | 2.00 | 54,583 | 7.74 | ||||||||||||||
|
$2.80 - $3.20 |
133,500 | 7.2 | 3.00 | 89,000 | 7.92 | |||||||||||||||
|
$3.40 - $5.20 |
134,468 | 8.96 | 5.12 | 51,134 | 8.99 | |||||||||||||||
|
$5.40 - $10.20 |
106,917 | 9.46 | 6.39 | 18,375 | 9.45 | |||||||||||||||
|
$10.40 - $34.00 |
50,150 | 4.87 | 24.18 | 50,150 | 4.87 | |||||||||||||||
| 480,035 | 8.21 | $ | 6.45 | 263,242 | 7.61 | |||||||||||||||
The cumulative grant date fair value of employee options vested during the years ended December 31, 2017 and 2016 was $63,000 and $122,000, respectively. Total proceeds received for options exercised during the years ended December 31, 2017 and 2016 were $7,000 and $0, respectively.
As of December 31, 2017 and 2016, total compensation expense related to unvested employee stock options not yet recognized was $551,000 and $353,000, respectively, which is expected to be allocated to expenses over a weighted-average period of 1.97 and 2.46 years, respectively.
The assumptions used in calculating the fair value under the Black-Scholes option valuation model are set forth in the following table for options issued by the Company for the years ended December 31, 2017 and 2016:
|
2017 |
2016 |
||
|
Common share fair value |
$0.26 – $0.42 |
$0.16 – 0.24 |
|
|
Risk-free interest rate |
1.1% |
0.8% |
|
|
Expected dividend yield |
0% |
0% |
|
|
Expected option life |
4.5 |
4.6 |
|
|
Expected stock price volatility |
84.7 – 156.8% |
92.0 – 185.1% |
Nonemployee share-based compensation
We account for stock options granted to nonemployees in accordance with FASB ASC 505 which requires, among other things, that the amount of compensation expense recorded is subject to periodic adjustment until the underlying options vest. In connection with stock options granted to nonemployees, we recorded $308,000 and $184,000 for nonemployee share-based compensation during the years ended December 31, 2017 and 2016, respectively. These amounts were based upon the fair values of the vested portion of the grants. Amounts expensed during the remaining vesting period will be determined based on the fair value at the time of vesting using the Black Scholes option valuation model.
|
13. |
Income Taxes |
We have incurred net operating losses since inception. We have not reflected the benefit of net operating loss carryforwards in the accompanying financial statements and have established a full valuation allowance against our deferred tax assets.
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes as well as operating losses and tax credit carryforwards.
The significant components of our deferred tax assets and liabilities are as follows (in thousands):
|
December 31, |
||||||||
|
2017 |
2016 |
|||||||
|
Deferred tax assets (liabilities): |
||||||||
|
Non-capital losses carried forward |
$ | 7,233 | $ | 6,917 | ||||
|
Research and development expenditures |
887 | 697 | ||||||
|
Share issue costs |
117 | 191 | ||||||
|
Patents and other |
319 | 211 | ||||||
|
Property and equipment |
(4 | ) | 1 | |||||
|
Total deferred tax asset, net |
8,552 | 8,017 | ||||||
|
Valuation allowance |
(8,552 | ) | (8,017 | ) | ||||
|
Net deferred tax asset |
$ | — | $ | — | ||||
Realization of the future tax benefits is dependent on our ability to generate sufficient taxable income within the carry-forward period. Because of our history of operating losses, management believes that the deferred tax assets arising from the above-mentioned future tax benefits are currently not likely to be realized and, accordingly, we have provided a full valuation allowance.
The reconciliation of the Canadian statutory income tax rate applied to the net loss for the year to the income tax expense is as follows:
|
Year Ended December 31, |
||||||||
|
2017 |
2016 |
|||||||
|
Statutory income tax rate |
27.0 | % | 27.0 | % | ||||
|
Income tax recovery based on statutory rate |
(1,160 | ) | (594 | ) | ||||
|
Share-based compensation |
110 | 70 | ||||||
|
Gain on revaluation of warrant liability |
(2 | ) | — | |||||
|
Australian research and development incentive |
314 | — | ||||||
|
Share issue costs |
(94 | ) | (88 | ) | ||||
|
Other |
298 | (280 | ) | |||||
|
Change in unrecognized temporary differences |
534 | 892 | ||||||
|
Income tax expense |
— | — | ||||||
Net operating losses and tax credit carryforwards as of December 31, 2017, are as follows:
|
Amount |
Expiration Years |
||||
|
Non-capital income tax losses, net |
$ | 29,943 |
Beginning 2026 |
||
|
Research and development expense carry forwards |
3,284 |
Indefinitely |
|||
|
Tax credits |
525 |
Beginning 2020 |
|||
The Company is subject to taxation in the Canada, the United States and Australia. Tax returns, since the inception of DiaMedica Therapeutics Inc. are subject to examinations by Canadian tax authorities and may change upon examination. Tax returns of DiaMedica USA, Inc., since its inception in 2012 and thereafter, are subject to examination by the U.S. federal and state tax authorities. Tax returns of DiaMedica Therapeutics Australia Pty Ltd., since its inception in 2016 and thereafter, are subject to examination by the Australian tax authorities.
|
14. |
Subsequent Events |
For the audited consolidated financial statements, management evaluated subsequent events through August 27, 2018, the date these consolidated financial statements were available to be issued.
For the interim condensed consolidated financial statements, management evaluated subsequent events through September 17, 2018, the date these condensed consolidated financial statements were available to be issued. After the original issuance of our interim condensed consolidated financial statements, we evaluated subsequent events through November 19, 2018.
Sale of common shares and stock purchase warrants
On March 29, 2018, the Company completed, in two tranches, a brokered and non-brokered private placement of 1,322,965 units at a price of $4.90 per unit for aggregate gross proceeds of approximately $6.3 million. Each unit consisted of one common share and one half of one common share purchase warrant. Each whole warrant entitles the holder to purchase one common share at a price of $7.00 at any time prior to expiry on March 19, 2020 and March 29, 2020 for Tranche 1 and Tranche 2, respectively. The warrants are subject to early expiry under certain conditions. The warrant expiry date can be accelerated at the option of the Company, in the event that the volume-weighted average trading price of the Company’s common shares exceeds $12.00 per common share for any 21 consecutive trading days. In connection with this offering, the Company paid aggregate finder’s fees of approximately $384,000 and issued 80,510 compensation warrants. Each compensation warrant entitles the holder to purchase one common share at $4.90 for a period of 2 years from the closing of this offering, subject to acceleration on the same terms as the common share purchase warrants.
Issuance of common shares on the exercise of stock purchase warrants
During February 2018, 121,256 common shares were issued on the exercise of warrants for gross proceeds of approximately $484,000.
Issuance of stock options
On April 17, 2018, the Compensation Committee of the Board of Directors awarded 166,800 stock options to various officers, directors and employees of the Company. The options were issued at CAD$11.20 per common share, the closing price of the Company’s common shares on the date of grant and have a ten-year term.
License and collaboration agreement with related party
On September 27, 2018, we entered into a license and collaboration agreement (the “License Agreement”) with Ahon Pharma, which grants Ahon Pharma exclusive rights to develop and commercialize DM199 for acute ischemic stroke in mainland China, Taiwan, Hong Kong S.A.R. and Macau S.A.R. Under the terms of the agreement, we are entitled to receive an upfront payment of $5.0 million, consisting of $500,000 due upon signing the License Agreement and $4.5 million upon regulatory clearance to initiate a clinical trial in China. We also have the potential to receive up to an additional $27.5 million in development and sales related milestones and up to approximately 10% royalties on net sales of DM199 in the licensed territories. All development, regulatory, sales, marketing, and commercial activities and associated costs in the licensed territories will be the sole responsibility of Ahon Pharma. The License Agreement may be terminated at any time by Ahon Pharma by providing 120 days written notice.
Ahon Pharma is a subsidiary of Shanghai Fosun Pharmaceutical (Group) co. Ltd. (“Fosun Pharma”) which, through its partnership with SK Group, a South Korea based company, is an investor in DiaMedica, holding approximately 12.7% of our common shares as of September 30, 2018. This investment was made in 2016.
Share Consolidation
Effective November 15, 2018, we implemented a 1-for-20 consolidation of our common shares. No fractional shares were issued in connection with the share consolidation. Instead, the Company rounded to the nearest whole number the number of shares shareholders would be entitled to receive in connection with the consolidation. The share consolidation was intended to increase the market price per share of our common shares to a level that qualifies for listing on The Nasdaq Capital Market. The Company is taking additional actions to meet the remaining listing requirements. Until the Company meets the criteria for listing and an application for listing is accepted by Nasdaq, which may not happen within a reasonable time frame, if at all, its common shares will continue to be listed on the TSX Venture Exchange and eligible for quotation on OTCQB Marketplace tier of the over-the-counter markets administered by the OTC Markets Group, Inc. under the symbol “DMCAF”. Proportional adjustments were also made to common shares reserved for issuance under the Company’s Option Plan and 2012 DSU Plan and outstanding stock options and warrants. The new CUSIP number for our common shares following the share consolidation is 25253X207. All references to share and per share amounts included in these consolidated financial statements have been retroactively restated to reflect the share consolidation.
DiaMedica Therapeutics Inc.
Condensed Consolidated Balance Sheets
(In thousands, except share amounts)
|
September 30, 2018 |
December 31, 2017 |
|||||||
|
|
(Unaudited) |
|||||||
| ASSETS | ||||||||
|
Current assets: |
||||||||
|
Cash |
$ | 3,898 | $ | 1,353 | ||||
|
Amounts receivable |
1,043 | 80 | ||||||
|
Prepaid expenses |
160 | 61 | ||||||
|
Total current assets |
5,101 | 1,494 | ||||||
|
Deposit |
271 | 271 | ||||||
|
Property and equipment, net |
91 | 37 | ||||||
|
Total non-current assets |
362 | 308 | ||||||
|
Total Assets |
$ | 5,463 | $ | 1,802 | ||||
|
LIABILITIES AND STOCKHOLDERS’ EQUITY |
||||||||
|
Current liabilities: |
||||||||
|
Accounts payable and accrued liabilities |
$ | 1,353 | $ | 919 | ||||
|
Warrant liability |
— | 84 | ||||||
|
Total current liabilities |
1,353 | 1,003 | ||||||
|
Shareholders’ equity: |
||||||||
|
Common shares, no par value; unlimited authorized; 7,839,176 and 6,370,663 shares issued and outstanding, as of September 30, 2018 and December 31, 2017, respectively |
— | — | ||||||
|
Additional paid-in capital |
48,116 | 41,033 | ||||||
|
Accumulated deficit |
(44,006 | ) | (40,234 | ) | ||||
|
Total stockholders’ equity |
4,110 | 799 | ||||||
|
Total liabilities and stockholders’ equity |
$ | 5,463 | $ | 1,802 | ||||
See accompanying notes to condensed consolidated financial statements.
DiaMedica Therapeutics Inc.
Condensed Consolidated Statements of Operations
(In thousands, except share and per share amounts)
(Unaudited)
|
Three Months Ended September 30, |
Nine Months Ended September 30, |
|||||||||||||||
|
2018 |
2017 |
2018 |
2017 |
|||||||||||||
|
Operating revenues |
||||||||||||||||
|
License revenues |
$ | 500 | $ | — | $ | 500 | $ | — | ||||||||
|
Operating expenses: |
||||||||||||||||
|
Research and development |
1,210 | 411 | 3,071 | 2,577 | ||||||||||||
|
General and administrative |
777 | 415 | 2,073 | 941 | ||||||||||||
|
Total operating expense |
1,987 | 826 | 5,144 | 3,518 | ||||||||||||
|
Operating loss |
(1,487 | ) | (826 | ) | (4,644 | ) | (3,518 | ) | ||||||||
|
Other (income) expense: |
||||||||||||||||
|
Governmental assistance - research incentives |
(196 | ) | (244 | ) | (1,046 | ) | (244 | ) | ||||||||
|
Other (income) expense |
40 | (16 | ) | 61 | 14 | |||||||||||
|
Change in fair value of warrant liability |
— | 141 | 39 | 208 | ||||||||||||
|
Total other (income) expense |
(157 | ) | (119 | ) | (946 | ) | (22 | ) | ||||||||
|
Loss before income tax expense |
$ | (1,330 | ) | $ | (707 | ) | $ | (3,698 | ) | $ | (3,496 | ) | ||||
|
Income tax expense |
57 | — | 74 | — | ||||||||||||
|
Net loss |
(1,387 | ) | (707 | ) | (3,772 | ) | (3,496 | ) | ||||||||
|
Basic and diluted net loss per share |
$ | (0.18 | ) | $ | (0.12 | ) | $ | (0.51 | ) | $ | (0.60 | ) | ||||
|
Weighted average shares outstanding – basic and diluted |
7,836,683 | 6,055,364 | 7,406,378 | 5,848,178 | ||||||||||||
See accompanying notes to the condensed consolidated financial statements.
DiaMedica Therapeutics Inc.
Condensed Consolidated Statements of Shareholders’ Equity
(In thousands except share and per share amounts)
(Unaudited)
|
Additional |
Total |
|||||||||||||||
|
Common |
Paid-In |
Accumulated |
Shareholders’ |
|||||||||||||
|
Shares |
Capital |
Deficit |
Equity |
|||||||||||||
|
Balances at December 31, 2017 |
6,370,663 | $ | 41,033 | $ | (40,234 | ) | $ | 799 | ||||||||
|
Issuance of common shares and warrants, net of offering costs of $529 |
1,322,965 | 5,840 | — | 5,840 | ||||||||||||
|
Exercise of common share purchase warrants |
128,594 | 645 | — | 645 | ||||||||||||
|
Exercise of stock options |
16,954 | 43 | — | 43 | ||||||||||||
|
Share-based compensation expense |
— | 555 | — | 555 | ||||||||||||
|
Net loss |
— | — | (3,772 | ) | (3,772 | ) | ||||||||||
|
Balances at September 30, 2018 |
7,839,176 | $ | 48,116 | $ | (44,006 | ) | $ | 4,110 | ||||||||
See accompanying notes to the condensed consolidated financial statements.
DiaMedica Therapeutics Inc.
Condensed Consolidated Statements of Cash Flows
(In thousands)
(Unaudited)
|
Nine Months Ended September 30, |
||||||||
|
2018 |
2017 |
|||||||
|
Cash flows from operating activities: |
||||||||
|
Net loss |
$ | (3,772 | ) | $ | (3,495 | ) | ||
|
Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||
|
Share-based compensation |
555 | 358 | ||||||
|
Change in fair value of warrant liability |
39 | 208 | ||||||
|
Depreciation |
10 | 2 | ||||||
|
Changes in operating assets and liabilities: |
||||||||
|
Amounts receivable |
(963 | ) | (10 | ) | ||||
|
Prepaid expenses |
(99 | ) | (5 | ) | ||||
|
Accounts payable and accrued liabilities |
434 | 54 | ||||||
|
Net cash used in operating activities |
(3,796 | ) | (2,888 | ) | ||||
|
Cash flows from investing activities: |
||||||||
|
Purchase of property and equipment |
(63 | ) | (7 | ) | ||||
|
Net cash used in financing activities |
(63 | ) | (7 | ) | ||||
|
Cash flows from financing activities: |
||||||||
|
Proceeds from issuance of common shares and warrants, net of offering costs |
5,840 | 1,983 | ||||||
|
Proceeds from the exercise of common share purchase warrants |
522 | — | ||||||
|
Proceeds from the exercise of stock options |
43 | 7 | ||||||
|
Net cash provided by financing activities |
6,404 | 1,990 | ||||||
|
Net increase (decrease) in cash |
2,545 | (905 | ) | |||||
|
Cash at beginning of period |
1,353 | 1,736 | ||||||
|
Cash at end of period |
$ | 3,898 | $ | 831 | ||||
|
Supplemental disclosure of non-cash financing activities: |
||||||||
|
Reclassification of warrant liability upon warrant exercise |
$ | 123 | $ | — | ||||
See accompanying notes to the condensed consolidated financial statements.
DiaMedica Therapeutics Inc.
Notes to Condensed Consolidated Financial Statements
1. Business
DiaMedica Therapeutics Inc. and its wholly-owned subsidiaries, DiaMedica USA, Inc. and DiaMedica Australia Pty Ltd. (collectively “we,” “us,” “our” and the “Company”), exist for the primary purpose of advancing the clinical and commercial development of a proprietary recombinant KLK1 protein for the treatment of neurological and kidney diseases with our primary focus on acute ischemic stroke and chronic kidney disease. The Company is a listed company governed under the Canada Business Corporations Act and our shares are publicly traded on the TSX Venture Exchange in Canada under the symbol “DMA” and the OTCQB in the United States under the symbol “DMCAF.”
2. Risks and Uncertainties
The Company operates in a highly regulated and competitive environment. The development, manufacturing and marketing of pharmaceutical products require approval from, and are subject to ongoing oversight by, the Food and Drug Administration (“FDA”) in the United States, the European Medicines Agency (“EMA”) in the European Union, and comparable agencies in other countries. Obtaining approval for a new pharmaceutical product is never certain, may take many years, and is normally expected to involve substantial expenditures.
As of September 30, 2018, we have incurred losses of $44.0 million since our inception in 2000. For the nine months ended September 30, 2018, we incurred a net loss and negative cash flows from operating activities of $3.8 million. We expect to incur substantial losses for the foreseeable future, which will continue to generate negative net cash flows from operating activities, as we continue to pursue increasing levels of research and development activities and the clinical development of our primary product candidate, DM199. As of September 30, 2018, we had cash of $3.9 million, working capital of $3.7 million and shareholders’ equity of $4.1 million. The Company’s principal sources of cash have included the issuance of equity securities.
The accompanying interim condensed consolidated financial statements have been prepared assuming that we will continue as a going concern which contemplates the realization of assets and liquidation of liabilities in the normal course of business and do not include any adjustments relating to the recoverability or classification of assets or the amounts of liabilities that might result from the outcome of these uncertainties. Our ability to continue as a going concern, realize the carrying value of our assets and discharge our liabilities in the ordinary course of business is dependent upon a number of factors, including our ability to obtain additional financing, the success of our development efforts, our ability to obtain marketing approval for our initial product candidate, DM199, in the United States, the European Union or other markets and ultimately our ability to market and sell our initial product candidate. These factors, among others, raise substantial doubt about our ability to continue operations as a going concern. See Note 3 titled “Liquidity, Management’s Plans and Going Concern.”
3. Liquidity, Management Plans and Going Concern
As of December 31, 2017 and September 30, 2018, the Company has an accumulated deficit of $40.2 million and $44.0 million, respectively, and the Company has not generated positive cash flow from operations since its inception.
Additional funding will be required to continue the Company’s research and development and other operating activities. In the next 12 months we will likely seek to raise additional funds through various sources, such as equity and debt financings, or through strategic collaborations and license agreements. We can give no assurances that we will be able to secure additional sources of funds to support our operations, or if such funds are available to us, that such additional financing will be sufficient to meet our needs or on terms acceptable to us. This risk would increase if our clinical data is not positive or economic and market conditions deteriorate.
During March 2018, the Company completed a brokered and non-brokered private placement of 1,322,965 units at a price of $4.90 per unit for aggregate gross proceeds of approximately $6.3 million. In addition, during the nine months ended September 30, 2018, 128,594 common shares were issued on the exercise of warrants and 16,954 common shares were issued on the exercise of stock options for gross proceeds of approximately $522,000 and 43,000, respectively.
On September 27, 2018, we entered into a license and collaboration agreement with Ahon Pharmaceutical Co. Ltd. (“Ahon Pharma”). Under the terms of the license agreement, we received an upfront payment of $500,000 on signing of the agreement. See Note 12 titled “License and Collaboration Agreement” for additional information.
If we are unable to obtain additional financing when needed, we would need to scale back our operations taking actions that may include, among other things, reducing use of outside professional service providers, reducing staff or staff compensation, significantly modify or delay the development of our DM199 product candidate, license to third parties the rights to commercialize our DM199 product candidate for acute ischemic stroke, chronic kidney disease or other applications that we would otherwise seek to pursue, or cease operations.
Our future success is dependent upon our ability to obtain additional financing, the success of our development efforts, our ability to demonstrate clinical progress for our DM199 product candidate in the United States or other markets, our ability to obtain required governmental approvals of our product candidate and ultimately our ability to license or market and sell our DM199 product candidate. If we are unable to obtain additional financing when needed, if our clinical trials are not successful, if we are unable to obtain required governmental approvals, we would not be able to continue as a going concern and would be forced to cease operations and liquidate our company.
There can be no assurances that we will be able to obtain additional financing on commercially reasonable terms, or at all. The sale of additional equity securities would likely result in dilution to our current shareholders.
4. Basis of presentation
We have prepared the accompanying interim condensed consolidated financial statements in accordance with accounting principles general accepted in the United States (“US GAAP”) for interim financial information and with the instructions to Form 10-Q and Regulation S-X of the Securities and Exchange Commission (“SEC”). Accordingly, they do not include all of the information and footnotes required by US GAAP for complete financial statements. These interim condensed consolidated financial statements reflect all adjustments consisting of normal recurring accruals, which, in the opinion of management, are necessary to present fairly our consolidated financial position, consolidated results of operations, consolidated statement of shareholders’ equity and consolidated cash flows for the periods and as of the dates presented. Our fiscal year ends on December 31. The condensed consolidated balance sheet as of December 31, 2017 was derived from audited consolidated financial statements but does not include all disclosures required by US GAAP. These interim condensed consolidated financial statements should be read in conjunction with the annual consolidated financial statements and the notes thereto. The nature of our business is such that the results of any interim period may not be indicative of the results to be expected for the entire year. Certain prior period amounts have been reclassified to conform to the current basis of presentation.
Effective November 15, 2018, we implemented a 1-for-20 consolidation of our common shares. As previously announced, the share consolidation was approved by our stockholders as of November 6, 2018 and was intended to increase the market price per share of our common shares to a level that qualifies for listing on the Nasdaq Capital Market. All references to share and per share amounts included in these condensed consolidated financial statements have been retroactively restated to reflect the share consolidation. See Note 14 titled “Subsequent Events” for additional information.
Recently adopted accounting pronouncements
In May 2014, the Financial Accounting Standards Board (FASB) issued a new accounting standard that amends the guidance for the recognition of revenue from contracts with customers to transfer goods and services. The FASB subsequently issued additional, clarifying standards to address issues arising from implementation of the new revenue recognition standard. The new revenue recognition standard and clarifying standards require an entity to recognize revenue when control of promised goods or services is transferred to the customer at an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. We adopted this new standard as of January 1, 2018, but the adoption as of this date had no impact on our financial statements, as we had no revenue until the third quarter of 2018. We followed ASC 606, “Revenue from Contracts with Customers” in accounting for our License and Collaboration agreement with Ahon Pharma (Note 12).
Recently issued accounting pronouncements
In February 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2016-02, Leases. The guidance in ASU 2016-02 supersedes the lease recognition requirements in the Accounting Standards Codification Topic 840, Leases. ASU 2016-02 requires an entity to recognize assets and liabilities arising from a lease for both financing and operating leases, along with additional qualitative and quantitative disclosures. The new standard requires the immediate recognition of all excess tax benefits and deficiencies in the income statement and requires classification of excess tax benefits as an operating activity as opposed to a financing activity in the statements of cash flows. ASU 2016-02 is effective for fiscal years beginning after December 15, 2018, with early adoption permitted. Management is evaluating the standard's impact on the consolidated financial statements.
In June 2018, the FASB issued ASU 2018-07, Compensation – Stock Compensation (Topic 718): Improvements to Nonemployee Share-Based Payment Accounting, which simplifies several aspects of the accounting for nonemployee share-based payment transactions resulting from expanding the scope of Topic 718 to include share-based payment transactions for acquiring goods and services from nonemployees. This ASU is effective for the Company for fiscal years beginning after December 15, 2018, including interim periods within that fiscal year. Early adoption is permitted, but no earlier than an entity’s adoption date of Topic 606. Management is currently evaluating the impact of the new guidance on our consolidated financial statements.
5. Summary of Significant Accounting Policies
Principles of consolidation
The accompanying interim condensed consolidated financial statements include the assets, liabilities and expenses of DiaMedica Therapeutics Inc., and our wholly-owned subsidiaries, DiaMedica USA, Inc. and DiaMedica Australia Pty Ltd. All significant intercompany transactions and balances have been eliminated in consolidation.
Functional currency
The United States dollar is the functional currency that represents the economic effects of the underlying transactions, events and conditions and various other factors including the currency of historical and future expenditures and the currency in which funds from financing activities are mostly generated by the Company. A change in the functional currency occurs only when there is a material change in the underlying transactions, events and condition. A change in functional currency could result in material differences in the amounts recorded in the consolidated statement of loss and comprehensive loss for foreign exchange gains and losses. All amounts in the accompanying condensed consolidated financial statements are in U.S. dollars unless otherwise indicated.
Use of estimates
The preparation of condensed consolidated financial statements requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the condensed consolidated financial statements and the reported amount of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Concentration of credit risk
Financial instruments that potentially subject the Company to significant concentrations of credit risk consist primarily of cash. Cash is deposited in demand accounts at commercial banks. At times, such deposits may be in excess of insured limits. The Company has not experienced any losses on its deposits of cash.
Fair value of financial instruments
Carrying amounts of certain of the Company’s financial instruments, including amounts receivable, prepaid expenses and other current assets, accounts payable and accrued liabilities approximate fair value due to their short maturities. Certain of the Company’s common share purchase warrants are required to be reported at fair value. The fair value of common share purchase warrants is disclosed in Note 10 titled “Warrant Liability.”
Fair value measurements
Fair value is defined as the exit price, or amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants as of the measurement date. The authoritative guidance also establishes a hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. Observable inputs are inputs market participants would use in valuing the asset or liability developed based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company’s assumptions about the factors market participants would use in valuing the asset or liability developed based upon the best information available in the circumstances. The categorization of financial assets and financial liabilities within the valuation hierarchy is based upon the lowest level of input that is significant to the fair value measurement.
The hierarchy is broken down into three levels defined as follows:
| ● | Level 1—Unadjusted quoted prices in active markets that are accessible at the measurement date for identical, unrestricted assets or liabilities. | |
| ● | Level 2—Quoted prices for similar assets and liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets that are not active or model-derived valuations for which all significant inputs are observable, either directly or indirectly. | |
| ● | Level 3—Prices or valuation techniques that require inputs that are both significant to the fair value measurement and unobservable. |
Our cash and equivalents consist of bank deposits. As of September 30, 2018, the Company believes that the carrying amounts of its other financial instruments, including amounts receivable, accounts payable and accrued liabilities, approximate their fair value due to the short-term maturities of these instruments.
Common share warrant liability
The common share warrants that were issued in connection with the February 2016 private placements of common shares are classified as a liability in the consolidated balance sheets, as the common share warrants have an exercise price stated in Canadian dollars, which is different than the functional currency, and thus these warrants qualify as a derivative instruments. The fair value of these common share warrants is re-measured at each financial reporting period and immediately before exercise, with any changes in fair value being recognized as a component of other income (expense) in the consolidated statements of operations. These warrants were exercised in February 2018, see Note 10 titled “Warrant Liability.”
Long-lived assets
Property and equipment are stated at purchased cost less accumulated depreciation. Depreciation of property and equipment is computed using the straight-line method over their estimated useful lives of three to ten years for office equipment and four years for computer equipment. Upon retirement or sale, the cost and related accumulated depreciation are removed from the consolidated balance sheets and the resulting gain or loss is reflected in the consolidated statements of operations. Repairs and maintenance are expensed as incurred.
Long-lived assets are evaluated for impairment when events or changes in circumstances indicate that the carrying amount of the asset or related group of assets may not be recoverable. If the expected future undiscounted cash flows are less than the carrying amount of the asset, an impairment loss is recognized at that time. Measurement of impairment may be based upon appraisal, market value of similar assets or discounted cash flows.
Revenue recognition
We followed ASC 606, “Revenue from Contracts with Customers” in accounting for our License and Collaboration agreement with Ahon Pharma. Accordingly, the Company recognizes revenue upon transfer of control of the product to our customer in an amount that reflects the consideration we expect to receive in exchange.
We intend to enter into arrangements for the research and development (R&D) and/or manufacture of products and product candidates. Such arrangements may require us to deliver various rights, services and/or goods, including (i) intellectual property rights or licenses, (ii) R&D services or (iii) manufacturing services. The underlying terms of these arrangements generally would provide for consideration to DiaMedica in the form of nonrefundable, up-front license fees, development and commercial-performance milestone payments, cost sharing, royalty payments and/or profit sharing.
In arrangements involving more than one performance obligation, each required performance obligation is evaluated to determine whether it qualifies as a distinct performance obligation based on whether (i) the customer can benefit from the good or service either on its own or together with other resources that are readily available and (ii) the good or service is separately identifiable from other promises in the contract. The consideration under the arrangement is then allocated to each separate distinct performance obligation based on its respective relative stand-alone selling price. The estimated selling price of each deliverable reflects our best estimate of what the selling price would be if the deliverable was regularly sold by us on a stand-alone basis or using an adjusted market assessment approach if selling price on a stand-alone basis is not available.
The consideration allocated to each distinct performance obligation is recognized as revenue when control of the related goods or services is transferred. Consideration associated with at-risk substantive performance milestones is recognized as revenue when it is probable that a significant reversal of the cumulative revenue recognized will not occur. We intend to utilize the sales and usage-based royalty exception in arrangements that resulted from the license of intellectual property, recognizing revenues generated from royalties or profit sharing as the underlying sales occur.
Research and development costs
Research and development costs include expenses incurred in the conduct of human clinical trials, for third-party service providers performing various testing and accumulating data related to non-clinical studies; sponsored research agreements; developing the manufacturing process necessary to produce sufficient amounts of the DM199 compound for use in our non-clinical studies and human clinical trials; consulting resources with specialized expertise related to execution of our development plan for our DM199 product candidate; and personnel costs, including salaries, benefits and share-based compensation.
We charge research and development costs, including clinical trial costs, to expense when incurred. Our human clinical trials are performed at clinical trial sites and are administered jointly by us with assistance from contract research organizations (“CROs”). Costs of setting up clinical trial sites are accrued upon execution of the study agreement. Expenses related to the performance of clinical trials are accrued based on contracted amounts and the achievement of agreed upon milestones, such as patient enrollment, patient follow-up, etc. We monitor levels of performance under each significant contract, including the extent of patient enrollment and other activities through communications with the clinical trial sites and CROs, and adjust the estimates, if required, on a quarterly basis so that clinical expenses reflect the actual work performed at each clinical trial site and by each CRO.
Share-based compensation
The cost of employee and non-employee services received in exchange for awards of equity instruments is measured and recognized based on the estimated grant date fair value of those awards. Compensation cost is recognized ratably using the straight-line attribution method over the vesting period, which is considered to be the requisite service period. We record forfeitures in the periods in which they occur.
The fair value of share-based awards is estimated at the date of grant using the Black-Scholes option pricing model. The determination of the fair value of share-based awards is affected by our share price, as well as assumptions regarding a number of complex and subjective variables. Risk free interest rates are based upon Canadian Government bond rates appropriate for the expected term of each award. Expected volatility rates are based on the on historical volatility equal to the expected life of the option. The assumed dividend yield is zero, as we do not expect to declare any dividends in the foreseeable future. The expected term of options is estimated considering the vesting period at the grant date, the life of the option and the average length of time similar grants have remained outstanding in the past.
Government assistance
Government assistance relating to research and development performed by DiaMedica Australia Pty Ltd. is recorded as a component of Other (income) expense. Government assistance is initially recognized when reasonable assurance exists that the Company will comply with the conditions attached to the incentive program and that the incentive payments will be received. In subsequent periods, the government assistance is recognized when the related expenditures are incurred.
Net loss per share
We compute net loss per share by dividing our net loss (the numerator) by the weighted-average number of common shares outstanding (the denominator) during the period. Shares issued during the period and shares reacquired during the period, if any, are weighted for the portion of the period that they were outstanding. The computation of diluted earnings per share, or EPS, is similar to the computation of basic EPS except that the denominator is increased to include the number of additional common shares that would have been outstanding if the dilutive potential common shares had been issued. Our diluted EPS is the same as basic EPS due to common equivalent shares being excluded from the calculation, as their effect is anti-dilutive.
The following table summarizes our calculation of net loss per common share for the periods (in thousands, except share and per share data):
|
Three Months Ended September 30, |
Nine Months Ended September 30, |
|||||||||||||||
|
2018 |
2017 |
2018 |
2017 |
|||||||||||||
|
Net loss |
$ | (1,387 | ) | $ | (707 | ) | $ | (3,772 | ) | $ | (3,496 | ) | ||||
|
Weighted average shares outstanding—basic and diluted |
7,836,683 | 6,055,364 | 7,406,378 | 5,848,178 | ||||||||||||
|
Basic and diluted net loss per share |
$ | (0.18 | ) | $ | (0.12 | ) | $ | (0.51 | ) | $ | (0.60 | ) | ||||
The following outstanding potential common shares were not included in the diluted net loss per share calculations as their effects were not dilutive:
|
Three Months Ended September 30, |
Nine Months Ended September 30, |
|||||||||||||||
|
2018 |
2017 |
2018 |
2017 |
|||||||||||||
|
Employee and non-employee stock options |
639,359 | 480,035 | 639,359 | 480,035 | ||||||||||||
|
Common shares issuable under common share purchase warrants |
825,264 | 391,260 | 825,264 | 391,260 | ||||||||||||
|
Common shares issuable under deferred share unit plan |
21,183 | 21,183 | 21,183 | 21,183 | ||||||||||||
6. Amounts Receivable
Amounts receivable consisted of the following (in thousands):
|
September 30, 2018 |
December 31, 2017 |
|||||||
|
Research and development incentives |
450 | — | ||||||
|
License fees |
455 | — | ||||||
|
Sales-based taxes receivable |
138 | 80 | ||||||
|
Total amounts receivable |
$ | 1,043 | $ | 80 | ||||
7. Deposit
Deposit consisted of the following (in thousands):
|
September 30, 2018 |
December 31, 2017 |
|||||||
|
Advances to vendor |
$ | 271 | $ | 271 | ||||
|
Total Deposit |
$ | 271 | $ | 271 | ||||
We have advanced funds to a vendor engaged to support the performance of the REMEDY Phase 2 clinical trial. The funds advanced will be held, interest free, by this vendor until the completion of the trial and applied to final trial invoices or refunded. This deposit is classified as non-current as the trial is not expected to be completed during 2018.
8. Property and Equipment
Property and equipment consisted of the following (in thousands):
|
September 30, 2018 |
December 31, 2017 |
|||||||
|
Furniture and equipment |
$ | 49 | $ | 40 | ||||
|
Computer equipment |
60 | 23 | ||||||
| 109 | 63 | |||||||
|
Less accumulated depreciation |
(18 | ) | (26 | ) | ||||
|
Property and equipment, net |
$ | 91 | $ | 37 | ||||
9. Accounts Payable and Accrued Liabilities
Accounts payable and accrued liabilities consisted of the following (in thousands):
|
September 30, 2018 |
December 31, 2017 |
|||||||
|
Trade and other payables |
$ | 249 | $ | 513 | ||||
|
Accrued compensation |
422 | 355 | ||||||
|
Accrued clinical study costs |
445 | — | ||||||
|
Accrued research and other professional fees |
184 | 45 | ||||||
|
Accrued taxes and other liabilities |
53 | 6 | ||||||
|
Total accrued liabilities |
$ | 1,353 | $ | 919 | ||||
10. Warrant Liability
In February 2016, the Company completed, in two tranches, a non-brokered private placement of 234,375 units with each unit consisting of one common share and one half of one common share purchase warrant. The Company issued 117,188 warrants. Each warrant entitles the holder to purchase one common share at a price of $5.00 Canadian dollars at any time prior to expiry on February 18 or 25, 2018 for Tranche 1 and Tranche 2, respectively.
As the warrant exercise price is stated in Canadian dollars and the Company’s functional currency is the U.S. dollar, the warrants are deemed to be a derivative, with their estimated fair value classified as a liability on the Company’s consolidated balance sheet. The initial estimated fair value of the warrants was recorded as a warrant liability with subsequent changes in the estimated fair value recognized in the consolidated statements of operations and comprehensive loss. The Company allocated $281,000 of the net proceeds to the warrant liability and the balance of the proceeds to the common shares (Note 9). The initial fair value of the warrants was determined using a Black- Scholes pricing model with the following assumptions: expected volatilities of 191.8 – 225.0%, risk-free interest rates of 0.43 – 0.49%, and expected life of 2 years.
In connection with the offering, the Company issued an aggregate of 10,915 compensation warrants. Each compensation warrant entitles the holder to purchase one common share at $5.00 Canadian dollars for a period of 2 years from the date of issuance, subject to acceleration on the same terms as the common share purchase warrants. The Company estimated the value of these warrants at $24,000, which was included in the issuance costs. The initial fair value of the warrants was determined using a Black-Scholes valuation model with the following assumptions: expected volatilities of 191.8 – 225.0%, risk-free interest rates of 0.43 – 0.49%, and expected life of 2 years.
During February 2018, 121,256 common shares were issued on the exercise of warrants for gross proceeds of approximately $483,000 and the remaining 4,346 warrants expired.
The fair value of the Company’s common share purchase warrant liability is calculated using a Black-Scholes valuation model and is classified as Level 3 in the fair value hierarchy. The fair values at the time of exercise of the warrants were estimated using the following valuation assumptions:
| Warrant Valuation | |||||
|
Common share fair value |
$0.31 | ||||
|
Risk-free interest rate |
1.84% | ||||
|
Expected dividend yield |
0% | ||||
|
Expected life (years) |
0.01 | – | 0.03 | ||
|
Expected share price volatility |
16.7% | ||||
The following is a rollforward of the fair value of Level 3 warrants (in thousands):
|
Warrant Liability |
||||
|
Ending balance December 31, 2017 |
$ | 84 | ||
|
Change in fair value |
39 | |||
|
Exercises |
(123 | ) | ||
|
Ending balance September 30, 2018 |
$ | — | ||
11. Shareholders’ Equity
Authorized capital stock
The Company has authorized share capital of an unlimited number of common voting shares and the shares do not have a stated par value.
Common shareholders are entitled to receive dividends as declared by the Company, if any, and are entitled to one vote per share at the Company's annual general meeting and any extraordinary general meeting.
Private placements during 2018
On March 29, 2018, the Company completed, in two tranches, a brokered and non-brokered private placement of 1,322,965 units at a price of $4.90 per unit for aggregate gross proceeds of approximately $6.3 million. Each unit consisted of one common share and one half of one common share purchase warrant. The Company issued 661,482 warrants. Each warrant entitles the holder to purchase one common share at a price of $7.00 at any time prior to expiry on March 19, 2020 and March 29, 2020 for Tranche 1 and Tranche 2, respectively. The warrants are subject to early expiry under certain conditions. The warrant expiry date can be accelerated at the option of the Company, in the event that the volume- weighted average trading price of the Company’s common shares exceeds $12.00 per common share for any 21 consecutive trading days. In connection with this offering, the Company paid aggregate finder’s fees of approximately $384,000 and issued an aggregate of 80,510 compensation warrants. Each compensation warrant entitles the holder to purchase one common share at $4.90 for a period of 2 years from the closing of this offering, subject to acceleration on the same terms as the common share purchase warrants.
During the nine months ended September 30, 2018, 128,594 common shares were issued on the exercise of warrants for gross proceeds of $665,000 and 16,954 common shares were issued on the exercise of options for gross proceeds of $43,000.
Private placements during 2017
On December 18, 2017, the Company completed a non-brokered private placement of 181,220 units at a price of $5.20 per unit for aggregate gross proceeds of approximately $944,000, or $934,000 net of issuance costs. Each unit consisted of one common share and one half of one common share purchase warrant. Each whole warrant entitles the holder to purchase one common share at a price of $7.00 at any time prior to expiry on December 19, 2019. Warrants are subject to early expiry, at the option of the Company, if on any date the volume-weighted average closing trading price of the common shares on any recognized Canadian
On April 17, 2017, the Company completed a non-brokered private placement of 526,316 units at a price of $3.80 per unit for aggregate gross proceeds of approximately $2,000,000, or $1,983,000 net of issuance costs. Each unit consisted of one common share and one half of one common share purchase warrant. Each whole warrant entitles the holder to purchase one common share at a price of $4.60 at any time prior to expiry on April 17, 2019. Warrants are subject to early expiry, at the option of the Company, if on any date the volume-weighted average closing trading price of the common shares on any recognized Canadian stock exchange equals or exceeds $6.00 for a period of 10 consecutive trading days.
During the year ended December 31, 2017, 2,500 common shares were issued on the exercise of warrants for gross proceeds of $9,913 and 3,000 common shares were issued on the exercise of options for gross proceeds of $6,749.
Shares reserved
Common shares reserved for future issuance are as follows:
|
September 30, 2018 |
||||
|
Stock options outstanding |
639,359 | |||
|
Deferred share units outstanding |
21,183 | |||
|
Shares available for grant under the DiaMedica Stock Option Plan |
123,376 | |||
|
Common shares issuable under common share purchase warrants |
825,264 | |||
|
Total |
1,609,182 | |||
12. License and Collaboration Agreement with Related Party
On September 27, 2018, we entered into a license and collaboration agreement (the “License Agreement”) with Ahon Pharma, which grants Ahon Pharma exclusive rights to develop and commercialize DM199 for acute ischemic stroke in mainland China, Taiwan, Hong Kong S.A.R. and Macau S.A.R. Under the terms of the agreement, we are entitled to receive a non-refundable upfront payment of $5.0 million, consisting of $500,000 due upon signing the License Agreement and $4.5 million upon regulatory clearance to initiate a clinical trial in China. We also have the potential to receive up to an additional $27.5 million in development and sales related milestones and up to approximately 10% royalties on net sales of DM199 in the licensed territories. All development, regulatory, sales, marketing, and commercial activities and associated costs in the licensed territories will be the sole responsibility of Ahon Pharma. The License Agreement may be terminated at any time by Ahon Pharma by providing 120 days written notice.
Ahon Pharma is a subsidiary of Shanghai Fosun Pharmaceutical (Group) co. Ltd. (“Fosun Pharma”) which, through its partnership with SK Group, a South Korea based company, is an investor in DiaMedica, holding approximately 12.7% of our common shares as of September 30, 2018. This investment was made in 2016.
We were entitled to receive the initial $500,000 license payment upon signing of the agreement and providing Ahon Pharma access to certain of our intellectual property rights and clinical data. There were no further performance obligations related to this payment. Accordingly, we recorded the license fee as revenue and an amount receivable as of September 30, 2018. Under the terms of the License Agreement, we are obligated to pay and Ahon Pharma may withhold, approximately 10% of any license fee as income tax due in China. We recorded this withholding as income tax at the time we recorded the related license fee revenue.
13. Share-Based Compensation
Deferred share unit plan
The 2012 Deferred Share Unit Plan (the “2012 DSU Plan”) promotes greater alignment of long-term interests between non-executive directors and executive officers of the Company and its shareholders through the issuance of deferred share units (“DSUs”). Since the value of DSUs increases or decreases with the market price of the common shares, DSUs reflect a philosophy of aligning the interests of directors and executive officers by tying compensation to share price performance. For the nine months ended September 30, 2018 and 2017, there were no DSUs or common shares underlying DSUs issued. The Company has reserved for issuance up to 100,000 common shares under the 2012 DSU Plan and 21,183 DSUs were outstanding at September 30, 2018.
Stock option plan
DiaMedica has adopted a Stock Option Plan (the “Option Plan”) where the Board of Directors may from time to time, in its sole discretion, and in accordance with the requirements of the TSX Venture Exchange, grant to directors, officers, management company employees, investor relations consultants and consultants (as such terms are used in the Stock Option Plan) to DiaMedica, non-transferable options to purchase common shares. The shareholders approved the adoption of the Option Plan on September 22, 2011, which was then amended and restated on October 23, 2015 and December 21, 2017, reserving for issuance up to 10% of the Company’s issued and outstanding common shares. Options granted vest at various rates and have terms of up to 10 years. As of September 30, 2018, options to purchase 639,359 common shares were outstanding. As the TSX Venture Exchange is the principal trading market for the Company’s shares, all options have been priced in Canadian dollars.
The aggregate number of common shares reserved as of September 30, 2018 was 783,918, which includes both the Option Plan and the 2012 DSU Plan.
Share-based compensation expense for each of the periods presented is as follows (in thousands):
|
Three Months Ended |
Nine Months Ended |
|||||||||||||||
|
September 30, 2018 |
September 30, 2017 |
September 30, 2018 |
September 30, 2017 |
|||||||||||||
|
Research and development |
$ | 26 | $ | 22 | $ | 129 | $ | 42 | ||||||||
|
General and administrative |
84 | 161 | 426 | 316 | ||||||||||||
|
Total share-based compensation |
$ | 110 | $ | 183 | $ | 555 | $ | 358 | ||||||||
We recognize share-based compensation based on the fair value of each award as estimated using the Black-Scholes option valuation model. Ultimately, the actual expense recognized over the vesting period will only be for those shares that actually vest.
A summary of option activity is as follows (in thousands except share and per share amounts):
|
Shares Underlying Options |
Weighted Average Exercise Price Per Share (CAD$) |
Aggregate Intrinsic Value (CAD$) |
||||||||||
|
Balances at December 31, 2017 |
480,034 | $ | 6.45 | $ | 674 | |||||||
|
Granted |
196,800 | 11.08 | ||||||||||
|
Exercised |
(16,954 | ) | 3.29 | |||||||||
|
Expired / cancelled |
(20,521 | ) | 8.99 | |||||||||
|
Balances at September 30, 2018 |
639,359 | $ | 7.87 | $ | 1,999 | |||||||
Information about stock options outstanding, vested and expected to vest as of September 30, 2018, is as follows:
|
Outstanding, Vested and Expected to Vest |
Options Vested and Exercisable |
|||||||||||||||||||||
|
Weighted Average |
Weighted |
Weighted Average |
||||||||||||||||||||
| Per Share |
Remaining |
Average |
Remaining |
|||||||||||||||||||
| Exercise Price |
Contractual |
Exercise Price |
Options |
Contractual |
||||||||||||||||||
| (CAD$) |
Shares |
Life (Years) |
(CAD$) |
Exercisable |
Life (Years) |
|||||||||||||||||
| $2.00 |
- |
$2.60 | 50,000 | 7.1 | $ | 2.00 | 50,000 | 7.1 | ||||||||||||||
| $2.80 |
- |
$3.20 | 125,400 | 7.2 | 3.00 | 114,275 | 7.2 | |||||||||||||||
| $3.40 |
- |
$5.20 | 130,405 | 8.2 | 5.12 | 81,551 | 8.2 | |||||||||||||||
| $5.40 |
- |
$10.20 | 98,063 | 8.7 | 6.39 | 42,646 | 8.7 | |||||||||||||||
| $10.40 |
- |
$34.00 | 235,491 | 8.4 | 13.86 | 64,050 | 5.3 | |||||||||||||||
| 639,359 | 8.1 | $ | 7.87 | 352,522 | 7.2 | |||||||||||||||||
The cumulative grant date fair value of employee options vested during the three months ended September 30, 2018 and 2017 was $81,000 and $55,000, respectively. The cumulative grant date fair value of employee options vested during the nine months ended September 30, 2018 and 2017 was $247,000 and $109,000, respectively.
Nonemployee share-based compensation
We account for stock options granted to nonemployees in accordance with FASB ASC 505. In connection with stock options granted to nonemployees, we recorded $239,000 and $247,000 for nonemployee share-based compensation during the nine months ended September 30, 2018 and 2017, respectively.
These amounts were based upon the fair values of the vested portion of the grants. Amounts expensed during the remaining vesting period will be determined based on the fair value at the time of vesting.
13. Subsequent Events
For the interim condensed consolidated financial statements, management evaluated subsequent events through November 19, 2018, the date these condensed consolidated financial statements were available to be issued.
Share consolidation
Effective November 15, 2018, we implemented a 1-for-20 consolidation of our common shares. No fractional shares were issued in connection with the share consolidation. Instead, the Company rounded to the nearest whole number the number of shares shareholders would be entitled to receive in connection with the consolidation. The share consolidation was intended to increase the market price per share of our common shares to a level that qualifies for listing on The Nasdaq Capital Market. The Company is taking additional actions to meet the remaining listing requirements. Until the Company meets the criteria for listing and an application for listing is accepted by Nasdaq, which may not happen within a reasonable time frame, if at all, its common shares will continue to be listed on the TSX Venture Exchange and eligible for quotation on OTCQB Marketplace tier of the over-the-counter markets administered by the OTC Markets Group, Inc. under the symbol “DMCAF”. Proportional adjustments were also made to common shares reserved for issuance under the Company’s Option Plan and 2012 DSU Plan and outstanding stock options and warrants. The new CUSIP number for our common shares following the share consolidation is 25253X207. All references to share and per share amounts included in these condensed consolidated financial statements have been retroactively restated to reflect the share consolidation.
Shares
Common Shares
________________________
PROSPECTUS
________________________
Craig-Hallum Capital Group
, 2018
Until , 2018 (25 days after the date of this prospectus), all dealers that effect transactions in these securities, whether or not participating in this offering, may be required to deliver a prospectus. This requirement is in addition to the dealers’ obligation to deliver a prospectus when acting as underwriter and with respect to their unsold allotments or subscriptions.
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution
The following table sets forth all costs and expenses, other than underwriting discounts and commissions, payable by us in connection with the sale of common shares being registered. All amounts shown are estimates except for the SEC registration fee, the FINRA filing fee and the Nasdaq listing fee.
|
SEC registration fee |
$ |
* |
|
FINRA filing fee |
$ |
* |
|
Nasdaq listing fee |
$ |
* |
|
Accountants’ fees and expenses |
$ |
* |
|
Legal fees and expenses |
$ |
* |
|
Blue sky fees and expenses |
$ |
* |
|
Transfer agent fees and expenses |
$ |
* |
|
Printing expenses |
$ |
* |
|
Miscellaneous |
$ |
* |
|
Total |
$ |
* |
________________________
|
* |
To be provided by amendment. |
Item 14. Indemnification of Directors and Officers
We are a corporation organized under the CBCA. Under Section 124 of the CBCA, a corporation may indemnify a present or former director or officer of the corporation or another individual who acts or acted at the corporation’s request as a director or officer, or an individual acting in a similar capacity, of another entity, against all costs, charges and expenses, including an amount paid to settle an action or satisfy a judgment, reasonably incurred by the individual in respect of any civil, criminal, administrative, investigative or other proceeding in which the individual is involved because of that association with the corporation or other entity. A corporation may not indemnify an individual unless the individual (i) acted honestly and in good faith with a view to the best interests of the corporation, or, as the case may be, to the best interests of the other entity for which the individual acted as a director or officer or in a similar capacity at the corporation's request, and (ii) in the case of a criminal or administrative action or proceeding that is enforced by a monetary penalty, the individual had reasonable grounds for believing that the conduct was lawful. Each of the aforementioned individuals are entitled to the indemnification provided above from a corporation as a matter of right if they were not judged by the court or other competent authority to have committed any fault or omitted to do anything that the individual ought to have done and if the individual fulfills conditions (i) and (ii) above. A corporation may advance moneys to a director, officer or other individual for the costs, charges and expenses of a proceeding; however, the individual shall repay the moneys if the individual does not fulfill the conditions set out in (i) and (ii) above. The indemnification or the advance of any moneys may be made in connection with a derivative action only with court approval and only if the conditions in (i) and (ii) above are met.
Under the CBCA, a corporation may purchase and maintain insurance for the benefit of any of the aforementioned individuals against any liability incurred by the individual in their capacity as a director or officer of the corporation, or in their capacity as a director or officer, or similar capacity, of another entity, if the individual acted in such capacity at the corporation’s request. We have maintained, and expect to continue to maintain, such an insurance policy covering our directors and officers with respect to certain liabilities.
We have entered into indemnification agreements with all of our directors and officers. The indemnification agreements are governed exclusively by and construed according to the substantive laws of the Canada, without regard to conflicts-of-laws principles that would require the application of any other law and provide, among other things, for indemnification to the fullest extent permitted by law and our by-laws against any and all expenses (including attorneys’ fees) and liabilities, judgments, fines and amounts paid in settlement that are paid or incurred by the executive or on his or her behalf in connection with such action, suit or proceeding. We will be obligated to pay these amounts only if the executive acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the best interests of our company. The indemnification agreements provide that the executive will not be indemnified and expenses advanced with respect to an action, suit or proceeding initiated by the executive unless (i) so authorized or consented to by our Board of Directors or the company has joined in such action, suit or proceeding or (ii) the action, suit or proceeding is one to enforce the executive’s rights under the indemnification agreement. Our indemnification and expense advance obligations are subject to the condition that an appropriate person or body not party to the particular action, suit or proceeding shall not have determined that the executive is not permitted to be indemnified under applicable law. The indemnification agreements also set forth procedures that apply in the event an executive requests indemnification or an expense advance.
Insofar as indemnification for liabilities arising under the Securities Act of 1933, as amended (the “Securities Act”), may be permitted to directors, officers or persons controlling us pursuant to the foregoing provisions, or otherwise, we have been advised that in the opinion of the Securities and Exchange Commission, such indemnification is against public policy as expressed in the Securities Act of 1933, as amended, and is, therefore, unenforceable.
Item 15. Recent Sales of Unregistered Securities
During the past three years, we issued unregistered securities as outlined below. Unless otherwise specifically noted, no commissions were paid in connection with the issuances described below and each issuance was effected pursuant to Section 4(a)(2) of the Securities Act, as a transaction by an issuer not involving any initial public offering.
During the period beginning on January 1, 2018 through November 15, 2018, we granted to certain of our employees, directors and consultants 196,800 stock options and no deferred share units (“DSUs”). During the year ended December 31, 2017, we granted to certain of our employees, directors and consultants 127,635 stock options and no DSUs. During the year ended December 31, 2016, we granted to certain of our employees, directors and consultants 138,750 stock options and 18,750 DSUs. During the year ended December 31, 2015, we granted to certain of our employees, directors and consultants 220,200 stock options and no DSUs. These securities were issued under our equity incentive plans without registration under the Securities Act in reliance on the exemptions afforded by Section 4(a)(2) of the Securities Act and Rule 701 promulgated thereunder.
Set forth below is information regarding additional securities issued by us within the past three years that were not registered under the Securities Act. The offers, sales and issuances of the securities described below were deemed to be exempt from registration under the Securities Act in reliance on Section 4(a)(2) (or Regulation D promulgated thereunder), in that the issuance of securities to the accredited investors did not involve a public offering. The recipients of securities in each of these transactions acquired the securities for investment only and not with a view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the securities issued in these transactions. Each of the recipients of securities in these transactions was an accredited investor under Rule 501 of Regulation D. No underwriters were involved in these transactions.
|
1. |
During the period beginning on January 1, 2018 and through November 15, 2018, 146,294 common shares were issued upon the exercise of warrants for gross proceeds of $758,000 and 16,954 common shares were issued upon the exercise of options for gross proceeds of $43,000. |
|
2. |
On March 29, 2018, we completed, in two tranches, a brokered and non-brokered private placement of 1,322,965 units at a price of $4.90 per unit for aggregate gross proceeds of approximately $6.3 million. Each unit consisted of one common share and one-half of one common share purchase warrant. Each whole warrant entitles the holder to purchase one common share at a price of $7.00 at any time prior to expiration on March 19, 2020 and March 29, 2020 for tranche 1 and tranche 2, respectively. The warrants are subject to early expiration under certain conditions. |
In connection with the offering, we paid an aggregate cash fee of approximately $384,000 to brokers and finders and issued 80,510 compensation options. Each compensation option entitles the holder to purchase one common share at $4.90, the offering price, for a period of two years from the closing of the offering, subject to acceleration on the same terms as the warrants issued to the investors.
|
3. |
On December 18, 2017, we completed a non-brokered private placement of 181,200 units at a price of $5.20 per unit for aggregate gross proceeds of approximately $944,000. Each unit consisted of one common share and one-half of one common share purchase warrant. Each whole warrant entitles the holder to purchase one common share at a price of $7.00 at any time prior to expiration on December 19, 2019, subject to early expiration under certain conditions. |
|
4. |
On April 17, 2017, we completed a non-brokered private placement of 526,316 units at a price of $3.80 per unit for aggregate proceeds of approximately $2,000,000. Each unit consists of one common share and one-half common share purchase warrant. Each whole warrant entitles the holder to purchase one common share at a price of $4.60 at any time prior to expiration on April 17, 2019. The warrant expiration date can be accelerated at our option in the event that the volume-weighted average trading price of our common shares exceeds $6.00 per common share for any 10 consecutive trading days. |
|
5. |
During the year ended December 31, 2017, 2,500 common shares were issued on the exercise of warrants for gross proceeds of $9,913 and 3,000 common shares were issued on the exercise of options for gross proceeds of $6,749. |
|
6. |
On September 8, 2016, we completed the second tranche of a non-brokered private placement of 750,000 common shares at a price of $4.00 per share for aggregate gross proceeds of $3,000,000. |
|
7. |
On August 22, 2016, we completed the first tranche of a non-brokered private placement of 250,000 common shares at a price of $4.00 per share for aggregate gross proceeds of $1,000,000. |
|
8. |
On April 22, 2016, we issued 2,500 common shares for settlement of a debt to a vendor at an issue price of $3.20 per common share. |
|
9. |
On February 25, 2016, we completed the second tranche of a non-brokered private placement of 190,625 units at a price of $2.34 per unit for aggregate gross proceeds of approximately $101,710. Each unit consisted of one common share and one-half of one common share purchase warrant. Each whole warrant entitled the holder to purchase one common share at a price of CAD$5.00 at any time prior to expiration of two years from the closing date. |
|
10. |
On February 18, 2016, we completed the first tranche of a non-brokered private placement of 190,625 units at a price of $2.34 per unit for aggregate gross proceeds of approximately $445,544. Each unit consisted of one common share and one-half of one common share purchase warrant. Each whole warrant entitled the holder to purchase one common share at a price of CAD$5.00 at any time prior to expiration of two years from the closing date. |
|
11. |
During the year ended December 31, 2016, 1,294 common shares were issued on the redemption of deferred share units and 174,108 common shares were issued on the exercise of warrants for gross proceeds of $442,000. |
|
12. |
On November 25, 2015, we announced the completion of a non-brokered private placement of 225,000 units at an issue price of $1.50 per unit for aggregate gross proceeds of $337,686. Each unit was comprised of one common share and one common share purchase warrant with each warrant entitling the holder thereof to acquire an additional common share at an exercise price of CAD$4.00 per share at any time prior to expiry on November 25, 2016. |
Item 16. Exhibits and Financial Statement Schedules
|
(a) |
Exhibits |
____________________
|
# |
Indicates a management contract or compensatory plan or arrangement. |
|
+ |
Previously filed. |
|
* |
Filed herewith. |
|
** |
To be filed by amendment. |
|
(1) |
Portions of this exhibit have been redacted and are subject to an application for confidential treatment under Rule 406 of the Securities Act of 1933, as amended. The redacted material was filed separately with the Securities and Exchange Commission. |
|
(b) |
Financial Statement Schedules |
All financial statement schedules are omitted because they are not applicable, the required information is not present in amounts sufficient to require submission of such schedules or the information is included in the financial statements or notes thereto.
Item 17. Undertakings
The undersigned registrant hereby undertakes to provide to the underwriter at the closing as specified in the underwriting agreement, certificates in such denominations and registered in such names as required by the underwriter to permit prompt delivery to each purchaser.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
The undersigned registrant hereby undertakes that:
(1) For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective.
(2) For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
Pursuant to the requirements of the Securities Act of 1933, the registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form S-1 and has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Minneapolis, State of Minnesota on November 19, 2018.
|
|
DIAMEDICA THERAPEUTICS INC. |
|
|
|
|
|
|
|
|
|
By |
/s/ Rick Pauls |
|
|
|
|
Rick Pauls |
|
|
|
|
President and Chief Executive Officer |
|
Pursuant to the requirements of the Securities Act of 1933, as amended, this registration statement has been signed by the following persons in the capacities and on the dates indicated:
|
Name and Signature |
Title |
Date |
||
| /s/ Rick Pauls |
President, Chief Executive Officer and Director (principal executive officer) |
November 19, 2018 | ||
|
Rick Pauls |
||||
| /s/ Scott Kellen |
Chief Financial Officer and Secretary |
November 19, 2018 | ||
|
Scott Kellen |
||||
| * |
Chairman of the Board |
November 19, 2018 | ||
|
Richard Pilnik |
||||
| * |
Director |
November 19, 2018 | ||
|
Michael Giuffre, M.D. |
||||
| * |
Director |
November 19, 2018 | ||
|
James Parsons |
||||
| * |
Director |
November 19, 2018 | ||
|
Zhenyu Xiao, Ph.D. |
* By: /s/ Rick Pauls
Rick Pauls
Attorney-in-Fact
II-6