
Exhibit 99.2
Updated Summary Business Information
Our Company
We are a clinical stage biopharmaceutical company primarily focused on the development of novel recombinant, or synthetic, proteins. Our goal is to use our patented and licensed technologies to establish our company as a leader in the development and commercialization of therapeutic treatments from novel recombinant proteins. Our current focus is on chronic kidney disease (CKD) and acute ischemic stroke (AIS). We plan to advance DM199, our lead drug candidate, through required clinical trials to create shareholder value by establishing its clinical and commercial potential as a therapy for CKD and AIS.
DM199 is a recombinant form of human tissue kallikrein-1 (KLK1). KLK1 is a serine protease (protein), produced primarily in the kidneys, pancreas and salivary glands, which plays a critical role in the regulation of local blood flow and vasodilation (the widening of blood vessels which decreases blood pressure) in the body, as well as an important role in inflammation and oxidative stress (an imbalance between potentially damaging reactive oxygen species, or free radicals, and antioxidants in the body). We believe DM199 has the potential to treat a variety of diseases where healthy functioning requires sufficient activity of KLK1 and its system, the kallikrein-kinin system (KKS).
CKD and AIS patients suffer from impaired blood flow to the kidneys and brain, respectively. These patients also tend to exhibit lower than normal levels of endogenous (produced by the body) KLK1. We believe treatment with DM199 could replenish levels of KLK1, thereby allowing the natural function of KKS to release bradykinin in the body where and when needed, generating beneficial nitric oxide and prostacyclin, setting in motion metabolic pathways that can improve blood flow (through vasoregulation), dampen inflammation and protect tissues and end-organs from ischemic damage, supporting structural integrity and normal functioning.
Today, forms of KLK1 derived from human urine and porcine pancreas are sold in Japan, China and Korea to treat AIS, CKD, retinopathy, hypertension and related vascular diseases. We believe millions of patients have been treated with these KLK1 therapies and the data from more than 100 published papers and studies support their clinical benefit. However, there are numerous regulatory, commercial and clinical drawbacks associated with KLK1 derived from human urine and porcine pancreas which can be overcome by developing a synthetic version of KLK1 such as DM199. We believe higher regulatory standards are the primary reason why KLK1 derived from human urine and porcine pancreas are not currently available and used in the United State or Europe. We are not aware of any synthetic version of KLK1 with regulatory approval for human use in any country, nor are we aware of any synthetic version in development other than our drug candidate, DM199.
As described in more detail below, positive top-line results from ReMEDy, a 92-subject study in acute ischemic stroke, including the achievement of primary safety and tolerability endpoints and no DM199-related serious adverse events, were announced in May 2020. In addition, there was also a demonstrated therapeutic effect in participants that received tissue plasminogen activator (tPA) prior to enrollment but not in participants receiving mechanical thrombectomy prior to enrollment according to top-line Phase II results.
We have conducted numerous internal and third-party analyses to evaluate the structural and functional performance of DM199 as compared to KLK1 derived from human urine. The results of these studies have demonstrated that DM199 is structurally and functionally equivalent to KLK1 derived from human urine in that (i) the amino acid structure of DM199 is identical to the human urine form, (ii) the enzymatic and pharmacokinetic profiles are substantially similar to human urinary derived KLK1 and (iii) the physiological effects of DM199 on blood pressure mirror that of human urinary derived KLK1. We believe that the results of this work suggest that the therapeutic action of DM199 will be the same or better than that of the forms of KLK1 marketed in Asia. In addition, we have completed enrollment in seven clinical trials with DM199 treating over 200 subjects, and the results have shown that DM199 has been safe and well-tolerated. However, DM199 has not been, and we cannot provide any assurance that it ultimately will be, determined to be safe or effective for purposes of granting marketing approval by the U.S. Food and Drug Administration (FDA) or any comparable agency.
Our recombinant form of DM199 is protected by issued composition of matter and delivery patents in the United States and Europe (expiration 2033); a pending worldwide patent (expiration 2038) that covers a range of DM199 dose levels and dosing regimens useful for treating a wide range of diseases associated with microvascular dysfunction; an exclusive license with our manufacturing partner for use of their cell line and proprietary expression system for manufacturing synthetic KLK1; and numerous trade-secrets. In addition, we believe DM199 cannot be reverse engineered to develop a copycat version of our therapy. This adds additional protection to our intellectual property, especially as we evaluate DM199 licensing.
Our Programs
The primary focus for our DM199 program development is currently on CKD and AIS. The current status of our product candidates in clinical development is as follows:
|
1. |
Due to the heightened risk of African Americans with the APOL1 genetic mutation to progress to end-stage renal disease, participants in this cohort will be tested for the presence of the APOL1 genetic mutation. |
|
2. |
Initiation of this cohort and development for this indication is contingent upon successful completion of this offering. |
Chronic Kidney Disease
CKD is a widespread health problem that generates significant economic burden throughout the world. According to the National Kidney Foundation, approximately 30 million Americans and 120 million Chinese suffer from this debilitating and potentially life-threatening condition. CKD is characterized by a progressive decline in overall kidney function, increasing the risk of premature death, cardiovascular events and hospitalization. End-stage renal disease (ESRD) is the final stage of CKD and requires ongoing dialysis or a kidney transplant to survive. However, many patients suffer serious health consequences or die from CKD prior to developing ESRD. Currently, there is no cure for CKD and treatment involves management of the symptoms of the disease. Blood pressure medications, such as angiotensin converting enzyme inhibitors (ACEi) or angiotensin receptor blockers (ARB), are often prescribed to control hypertension, and hopefully, slow the progression of CKD. Nevertheless, according to the National Kidney Foundation, many of these patients continue to show declining kidney function. We believe DM199 offers a potentially novel approach for the treatment of CKD because KLK1 protein plays a vital role in normal kidney function. Since patients with moderate to severe CKD often excrete abnormally low levels of KLK1 in their urine, we believe that DM199 may prevent or reduce further kidney damage by increasing levels of KLK1 and restoring the protective KKS to regulate the production and release of nitric oxide and prostacyclin.
Acute Ischemic Stroke
According to the World Health Organization, each year approximately 15 million people worldwide suffer a stroke, of which 5.0 million will die and 5.0 million will be permanently disabled. According to the U.S. Center for Disease Control and Prevention approximately 87% of all strokes are ischemic in nature, a blockage of blood flow in/to the brain. We believe that stroke represents an area of significant unmet medical need and a KLK1 treatment (such as DM199) could provide a significant patient benefit, in particular given its proposed therapeutic window of up to 24 hours after the first sign of symptoms. Currently, the only FDA-approved pharmacological intervention for AIS is tissue plasminogen activator (tPA), which must be given within 4.5 hours of symptom onset. Treating patients with tPA during this time window can be challenging because it is difficult to determine precisely when symptoms began and a patient must undergo complex brain imaging before treatment to rule out a hemorrhagic stroke, a ruptured blood vessel causing bleeding within the brain. Mechanical thrombectomy, a procedure in which the clot is removed using catheter-based tools, is also available to certain patients. Despite the availability of these treatments, we believe they are relevant to approximately 10% of ischemic stroke patients due to the location of the clot, the elapsed time after the stroke occurred or other safety considerations. Thus, we believe DM199 may offer significant advantages over the current treatment options in that it fills a serious, unmet need for patients who cannot receive tPA or mechanical thrombectomy. Additionally, DM199 may also offer a complimentary follow-on treatment for patients who initially receive tPA or mechanical thrombectomy treatments by enabling sustained blood flow improvements to the brain during the critical weeks and months after a stroke. Based on the number of strokes each year (approximately 1.7 million in the U.S., Europe and Japan and 15 million worldwide) and considering the $8,500 estimated cost per patient for the current standard of care, tPA, we believe the annual market opportunity for DM199 could be significant.
Our Clinical Trials
Chronic Kidney Disease
In July 2019, we completed a Phase Ib clinical trial of DM199 in participants with moderate or severe CKD caused by Type I or Type II diabetes. We initiated dosing patients in this study in February 2019. The study was performed to assess the pharmacokinetics (PK) of three dose levels of DM199 (3, 5 and 8 µg/kg), administered in a single subcutaneous dose, as well as the evaluation of safety, tolerability and secondary pharmacodynamic (PD) endpoints. The study results demonstrated that at the 3µg/kg dose level, the PK profiles were similar between moderate and severe CKD patients, and consistent with healthy subjects (normal kidney function) tested previously. Additionally, DM199 was well tolerated with no dose-limiting tolerability observed. There were no deaths, no discontinuations due to a treatment-related adverse event (AE) and no treatment-related significant adverse events (SAEs). AEs were minor and consistent with standard treatment(s) in the CKD patient population. We announced favorable overall interim PD results from the first 28 subjects that included short-term improvements in Nitric Oxide (NO), average increase of 35.2%, Prostaglandin E2 (PGE2), average increase of 41.2%, estimated glomerular flow rate (eGFR), average increase of 4.08 mL/min/1732, and the urinary albumin to creatinine ratio (UACR) excluding subjects with normal UACR levels, average decrease of 18.7%. PD results appeared to be drug related in that the greatest improvements occurred approximately 24 hours after DM199 administration and subsequently declined.
In December 2019, we began enrolling patients in a Phase II CKD trial named REDUX, Latin for restore, a multi-center, open-label investigation of approximately 60 participants with CKD, who are being enrolled in two cohorts (30 per cohort). The study is being conducted in the United States at up to 10 sites and will be focused on participants with two specific causes of CKD. Cohort I of the study is focused on non-diabetic, hypertensive African Americans with Stage II or III CKD. African Americans are at greater risk for CKD than Caucasians, and those who have the APOL1 gene mutation are at an even higher risk. The study is designed to capture the APOL1 gene mutation as an exploratory biomarker in this cohort. Cohort II of the study is focused on participants with IgA Nephropathy (IgAN). The study will evaluate two dose levels of DM199 within each cohort. Study participants will receive DM199 by subcutaneous injection twice weekly for 95 days. The primary study endpoints include safety, tolerability, blood pressure, albuminuria and kidney function, which will be evaluated by changes from baseline in eGFR and albuminuria, as measured by the UACR.
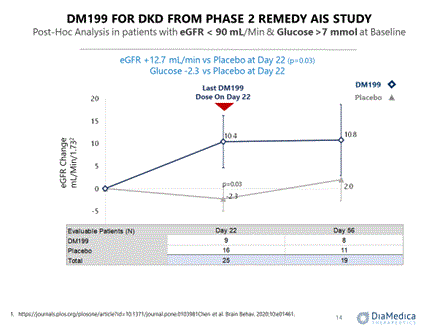
We intend to use a portion of the proceeds from this offering to begin enrollment in a third cohort in the REDUX trial comprised of participants with Type II diabetes mellitus with CKD, hypertension and albuminuria. In a post hoc analysis of endpoints in the ReMEDy trial, discussed below, a sub-set of participants with elevated blood glucose levels (>7 mmol/l) and impaired kidney function (eGFR <90) were observed to experience significant (mean +12.7 mL/min, p=0.03) improvement in kidney function as measured by the estimated glomerular filtration rate compared to placebo and a trending reduction in blood glucose levels (mean 2.2 mmol/l) compared to placebo. Initiation of this third cohort and development for this indication is contingent upon successful completion of this offering.
As of August 5, 2020, we have enrolled 18 subjects, including 7 African American subjects into cohort I and 11 subjects with IgAN into cohort II of the REDUX study. Due to actions implemented to combat the novel strain of the coronavirus (COVID-19) pandemic, we have experienced and continue to experience slower than expected enrollment in the REDUX clinical trial. We believe this is due to the reduction or suspension of activities at our clinical study sites as they address staff and patient safety concerns and patient concerns related to visiting clinical study sites in light of the pandemic. We anticipate that the COVID-19 pandemic will likely continue to adversely affect our ability to recruit or enroll subjects and we cannot provide any assurance as to when clinical sites will be able to resume enrollment at a normal rate or any guidance at this time as to when we will complete enrollment in the study. While results observed to date in the REDUX study indicate a safety profile consistent with past studies, there is insufficient data at this time to evaluate or comment upon efficacy.
Acute Ischemic Stroke
In May 2020, we announced top-line data from our Phase II ReMEDy trial assessing the safety, tolerability and markers of therapeutic efficacy of DM199 in patients suffering from AIS. We initiated treatment in this study in February 2018 and completed enrollment in October 2019 with 92 participants. The study drug (DM199 or placebo) was administered as an intravenous (IV) infusion within 24 hours of stroke symptom onset, followed by subcutaneous injections later that day and once every 3 days for 21 days. The study was designed to measure safety and tolerability along with multiple tests designed to investigate DM199’s therapeutic potential including plasma-based biomarkers and standard functional stroke measures assessed at 90 days post-stroke. Standard functional stroke measurements include the Modified Rankin Scale, National Institutes of Health Stroke Scale, the Barthel Index and C-reactive protein, a measure of inflammation. The study met primary safety and tolerability endpoints and there were no DM199-related serious adverse events. In addition, there was also a demonstrated therapeutic effect in participants that received tPA prior to enrollment but not in participants receiving mechanical thrombectomy prior to enrollment.
Prior to enrollment, 44 of the 91 evaluable patients (48%) received a mechanical thrombectomy, a catheter-based treatment indicated for those who have a large vessel occlusion and can be treated within 6 to 24 hours of the onset of stroke symptoms. While approximately 20% of AIS patients are believed to be eligible for a mechanical thrombectomy, currently only about 5% to 10% receive the treatment due to elapsed time post-stroke or unavailability of the therapy at the hospital where they present. DM199 is intended to treat the approximately 90% of AIS patients who do not receive either mechanical thrombectomy or tPA. Treatment for these patients is limited to palliative therapies. Due to the large volume of participants receiving mechanical thrombectomy prior to enrollment in ReMEDy, and a disproportionate distribution of these participants between the active treatment and placebo groups, DM199 did not produce a therapeutic effect in the overall study analysis.
When participants treated with mechanical thrombectomy are excluded from the study data set, representing the group of participants most closely aligned with the target treatment population for DM199, a positive therapeutic effect was demonstrated. As shown in the table below, when evaluating the participants treated with DM199 (n=25) vs. palliative therapies and/or tPA (n=21), the results showed that 36% of participants receiving DM199 progressed to a full or nearly full recovery at 90 days (NIHSS: 0-1), compared to 14% of participants in the placebo group. This represents a 22% absolute increase in the proportion of participants achieving a full or nearly full recovery. Additionally, subject deaths decreased from 24% in the placebo group to 12% in the active therapy group, a 50% relative reduction.
DM199 vs. Palliative Therapies and/or tPA
|
NIHSS Outcomes at 90 Days |
||||
|
0-1 |
2-8 |
≥ 9 |
Death |
|
|
Placebo (n=21) |
14% |
57% |
5% |
24% |
|
DM199 (n=24) |
36% |
36% |
16% |
12% |
In addition, in the evaluable participants (n=91), a significant reduction in the number of participants with severe recurrent stroke was noted in the active treatment group: 1 (2%) patient treated with DM199 vs. 7 (16%) on placebo (p=0.028), with 4 of the 7 resulting in participant death.
Further, in reviewing evaluable participants (n=91), improvements in the following biomarkers were observed in participants treated with DM199, which we believe are consistent with the DM199 mechanism of action:
|
● |
Increased NO (+105%) and PGE2 (+54%) were observed at day 22 vs baseline (p<0.05). Placebo group was not statistically significant vs baseline (p>0.05). These changes noted in the active treatment group did not reach statistical significance compared to placebo. |
|
● |
Reduction in C-reactive protein (CRP) of (-70%), a blood marker of inflammation, at 90 days. CRP decreased significantly vs. baseline (p<0.05), but was not statistically significant vs. placebo. The change in the placebo group was not statistically significant vs. baseline (p>0.05). |
|
● |
Reduction in elevated glucose levels in participants with type 2 diabetes, as defined by a blood glucose level >7 mmol/l (n=14), an average decrease of 1.9 mmol/l (p=0.06) in blood glucose levels of participants on active therapy was observed at day 22. In comparison, participants in the placebo group (n=16) showed an average increase of 0.08 mmol/l (p=0.94) at day 22. |
Changes in the eGFR, a measure of kidney function, were also analyzed in participants with eGFR <70 mL/Min/1.732 at baseline, which indicates the presence of CKD. Participants receiving DM199 exhibited a marked increase in eGFR at days 22 (last dose) and 56 (34 days post-treatment), as shown in the table below. eGFR at day 22 increased by at least 2 mL/Min in 77% of DM199 participants compared to 20% in placebo (p=0.007).
|
eGFR Mean Δ from Baseline (mL/Min/1.732) |
||
|
Day 22 (Last Dose) |
Day 56 (Off Treatment) |
|
|
Placebo |
+0.84 (n=15) |
-0.24 (n=12) |
|
DM199 |
+7.5 (n=13) |
+5.8 (n=12) |
|
Group Difference |
+6.6 |
+6.1 |
We believe these findings from our Phase II ReMEDy trial,which are consistent with Chinese data on the urine-derived form of KLK1, provide a signal that recombinant human KLK1 appears safe and may have promise as a new tool for physicians who have limited options for the treatment of patients suffering AIS and may also mitigate the adverse impact of ischemic stroke on kidney function.
Potential DM199 Commercial Advantages
The growing understanding of KLK1’s role in human health and its use in Asia as an approved therapeutic treatment highlights two important potential commercial advantages for DM199:
|
● |
KLK1 treatments currently sold in Japan, China and Korea. Research has shown that patients with low levels of KLK1 are associated with a variety of diseases related to vascular dysfunction, such as CKD, AIS, retinopathy and hypertension. Clinical trial data with human urine and porcine derived KLK1 has demonstrated statistically significant clinical benefits in treating a variety of patients with KLK1 compared to placebo. These efficacy results are further substantiated by established markets in Japan, China and Korea for pharmaceutical sales of KLK1 derived from human urine and porcine pancreas. We estimate that millions of patients have been treated with these forms of KLK1 in Asia. Altogether, we believe this supports a strong market opportunity for a synthetic version of KLK1 such as DM199. |
|
● |
KLK1 treatment has had limited side effects and has been well tolerated to date. KLK1 is naturally produced by the human body; and, therefore, the body’s own control mechanisms act to limit potential side effects. The only notable side effect observed in our clinical trials was orthostatic hypotension, or a sudden drop in blood pressure, which was only seen at doses ten to twenty times higher than our anticipated therapeutic dose levels. Moreover, routine clinical use of KLK1 treatment in Asia we understand has been well-tolerated by patients for several decades. In 2017, we completed a clinical trial comparing the pharmacokinetic profile of DM199 to the human urinary form of KLK1 (Kailikang), which showed DM199, when administered in intravenous form, had a similar pharmacokinetic profile. Further, when DM199 was administered subcutaneously, DM199 demonstrated a longer acting pharmacokinetic profile, superior to the intravenously administered Kailikang and DM199. |
In addition, we believe there are also significant formulation, manufacturing, regulatory and other advantages for our synthetic human KLK1 drug candidate DM199:
|
● |
Potency and Impurity Considerations. KLK1 derived from human urine or porcine pancreas may contain impurities, endotoxins, and chemical byproducts due to the inherent variability of the isolation and purification process. We believe that this creates the risk of inconsistencies in potency and impurities from one production run to the next. However, we expect to produce a consistent formulation of KLK1 that is free of endotoxins and other impurities. |
|
● |
Cost and Scalability. Large quantities of human urine and porcine pancreas must be obtained to derive a small amount of KLK1. This creates potential procurement, cost and logistical challenges to source the necessary raw material, particularly for human urine sourced KLK1. Once sourced, the raw material is processed using chemicals and costly capital equipment and produces a significant amount of byproduct waste. Our novel recombinant manufacturing process utilizes widely available raw materials and can be readily scaled for commercial production. Accordingly, we believe our manufacturing process will have significant cost and scalability advantages. |
|
● |
Regulatory. We are not aware of any attempts by manufacturers of the urine or porcine based KLK1 products to pursue regulatory approvals in the United States. We believe that this is related to challenges presented by using inconsistent and potentially hazardous biomaterials, such as human urine and porcine pancreas, and their resulting ability to produce a consistent drug product. Our novel recombinant manufacturing process utilizes widely available raw materials which we believe provides a significant regulatory advantage, particularly in regions such as the United States, Europe and Canada, where safety standards are high. In addition, we believe that DM199 could qualify for 12 years of data exclusivity under the Biologics Price Competition and Innovation Act of 2009, which was enacted as part of the Patient Protection and Affordable Care Act as amended by the Health Care and Education Reconciliation Act of 2010. |
Our Strategy
We aim to become a leader in the discovery, development and commercialization of recombinant proteins for the treatment of severe and life-threatening diseases. To achieve this goal, we are pursuing the following strategies:
|
● |
Complete our ongoing Phase II studies for DM199 in CKD patients; |
|
● |
Commence a Phase III study for DM199 in AIS patients; |
|
● |
Explore potential new indications for DM199; and |
|
● |
Leverage our experience and technologies to develop new recombinant therapies and programs. |
Our Team
We have assembled a seasoned management team with extensive experience in drug discovery, development and manufacturing. Our Chief Executive Officer, Rick Pauls, MBA, is a successful venture capitalist and formerly the Co-Founder and Managing Director of CentreStone Ventures Inc., a life sciences venture capital fund which made early investments in DiaMedica. Our Chief Medical Officer, Harry Alcorn Jr., Pharm. D, has more than 30 years’ experience planning, operating, and executing clinical development programs across a range of diseases including kidney disease, diabetes, and cardiovascular disease, and most recently served as Chief Scientific Officer of DaVita Clinical Research. Our Vice President, Regulatory Affairs, Sydney Gilman, Ph.D., has more than 30 years’ experience in drug research, regulatory affairs and quality assurance, including six years as a chemistry reviewer in FDA’s Center for Drug Evaluation and Research. Edward Calamai, our consulting head of manufacturing, has over 30 years’ experience guiding manufacturing operations, including senior positions at Sensu and Seragen. Dr. Calamai is currently the Managing Partner at PM&C Associates, a company he co-founded in 2001. Our Chief Financial Officer, Scott Kellen, CPA, brings over two decades of operational and corporate finance expertise including an extensive background working with publicly-traded healthcare and biotechnology companies.